Aus einer Forschungsarbeit von Jenny Do, Cindy McKinney, Pankaj Sharma & Ellen Sidransky
veröffentlicht in Molecular Neurodegeneration volume 14, Article number: 36 (2019)
Abstrakt
Mutationen im GBA1, dem Gen für das lysosomale Enzym Glukozerebrosidase, gehören zu den häufigsten bekannten genetischen Risikofaktoren für die Entwicklung der Parkinson-Krankheit und verwandter Synukleinopathien. Über GBA1 ist sehr viel bekannt, da Mutationen in GBA1 kausal für die seltene autosomale Speicherkrankheit Gaucher-Krankheit sind. In den letzten Jahrzehnten wurden bedeutende Fortschritte im Verständnis der Genetik und Zellbiologie der Glukozerebrosidase erzielt. Es wird von mindestens 495 verschiedenen Mutationen berichtet, die in den 11 Exons des Gens gefunden wurden, darunter sowohl häufige als auch seltene Varianten. Mutationen in GBA1 können zum Abbau des Proteins, zu Störungen des lysosomalen Targeting und zu einer verminderten Leistung des Enzyms im Lysosom führen.
Die Gaucher-Krankheit ist phänotypisch vielfältig und hat sowohl neuronopathische als auch nicht-neuronopathische Formen. Sowohl Patienten mit Morbus Gaucher als auch heterozygote Träger haben ein erhöhtes Risiko, an Parkinson und Demenz mit Lewy-Körpern zu erkranken, obwohl unser Verständnis des Mechanismus für diese Assoziation noch unvollständig ist. Es scheint eine umgekehrte Beziehung zwischen der Glukozerebrosidase und den α-Synuclein-Spiegeln zu bestehen, und selbst Patienten mit sporadischer Parkinson-Erkrankung haben eine verminderte Glukozerebrosidase. Die Glucocerebrosidase kann mit α-Synuclein interagieren, um grundlegende Zellfunktionen aufrechtzuerhalten, oder die beeinträchtigte Glucocerebrosidase könnte zur Parkinson-Pathogenese beitragen, indem sie die lysosomale Homöostase stört, den Stress des endoplasmatischen Retikulums verstärkt oder zu einer mitochondrialen Beeinträchtigung beiträgt.
Die Mehrheit der Patienten mit GBA1-Mutationen entwickelt jedoch nie Parkinson, so dass eindeutig andere Risikofaktoren eine Rolle spielen.
Es wurden Behandlungen für die Gaucher-Krankheit entwickelt, die die viszeralen Glukozerebrosidase-Werte erhöhen und die Lipidspeicherung verringern, obwohl sie die neurologischen Defekte, die mit der beeinträchtigten Glukozerebrosidase verbunden sind, noch nicht richtig angegangen sind. Modelle, die von der Maus und induzierten pluripotenten Stammzellen abgeleitet sind, haben unser Verständnis der Funktion der Glukozerebrosidase und der Folgen ihres Mangels verbessert. Diese Modelle wurden verwendet, um neue Therapien zu testen, darunter Chaperonproteine, Histondeacetylase-Inhibitoren und gentherapeutische Ansätze, die die Glukozerebrosidase-Werte erhöhen und sich bei der Behandlung von Formen des Parkinsonismus als wirksam erweisen könnten. Folglich bietet diese seltene monogene Erkrankung, die Gaucher-Krankheit, einzigartige Erkenntnisse, die direkt auf unser Verständnis und unsere Behandlung der Parkinson-Krankheit, einer häufigen und komplexen neurodegenerativen Erkrankung, anwendbar sind.
Hintergrund
Von allen bekannten genetischen Varianten, die mit der Parkinson-Krankheit assoziiert sind, haben Mutationen im GBA1, dem Gen, das für das lysosomale Enzym Glukozerebrosidase (Glucosylceramidase Beta oder GCase; EC 3.2.1.45) kodiert, aufgrund der Assoziation dieses Gens mit einer gut untersuchten lysosomalen Speicherkrankheit, der Gaucher-Krankheit, einen großen Vorteil. Die Gaucher-Krankheit, eine autosomal rezessiv vererbte Störung mit verschiedenen klinischen Manifestationen, wurde erstmals vor über 135 Jahren in Paris von einem Medizinstudenten, Philippe Gaucher, beschrieben, der einen Patienten mit einer massiv vergrößerten Milz untersuchte [1]. Erst ein halbes Jahrhundert später wurde entdeckt, dass es sich bei dem bei Patienten mit dieser Erkrankung gefundenen Speichermaterial tatsächlich um ein Glykolipid, das Glucosylceramid (GlcCer), handelt [2]. 1965 stellte Dr. Roscoe Brady an den National Institutes of Health in Bethesda, Maryland, fest, dass die Gaucher-Krankheit auf einen enzymatischen Defekt im lysosomalen Enzym Glucocerebrosidase (GCase) zurückzuführen ist, das normalerweise eine Glucose-Einheit von GlcCer abspaltet [1, 3]. Dieser Befund erleichterte die Reinigung des Proteins GCase, die Klonierung des GBA1-Gens 1981 und die Entwicklung der Enzymersatztherapie (ERT) als Behandlung von Patienten mit Morbus Gaucher [4]. In der Tat konzentrierte sich in den letzten Jahrzehnten viel Arbeit auf Mutationen im GBA1 und deren phänotypische Folgen. So ist im Gegensatz zu anderen neu entdeckten Parkinson-Genen viel über GBA1 und die Funktion des daraus resultierenden Enzyms GCase bekannt.
Glucocerebrosidase: Biochemie und Molekularbiologie
GCase ist ein 497-Aminosäure-Membran-assoziiertes Protein mit einer 39-Aminosäure-Leadersequenz und fünf Glykosylierungsstellen [4, 5]. Das Protein wird im endoplasmatischen Retikulum (ER) synthetisiert und glykosyliert, aber das Enzym wird erst aktiv, wenn es in das saure Lumen des Lysosoms übertragen wird (Abb. 1).
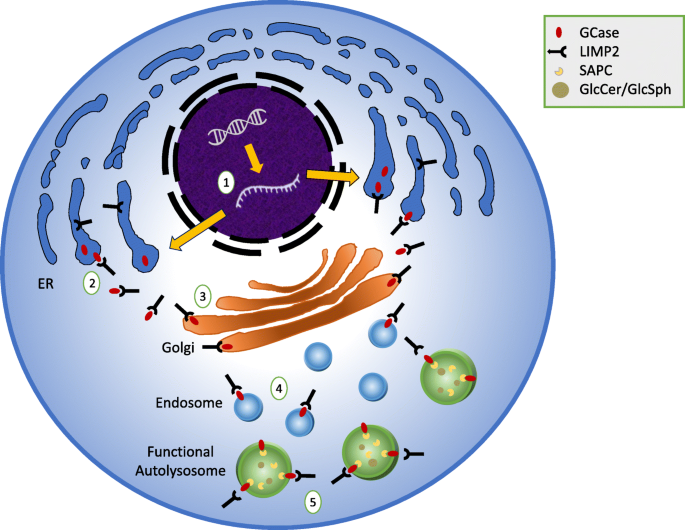
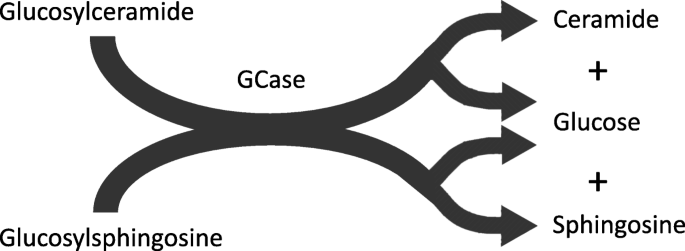
Im Gegensatz zu anderen lysosomalen Proteinen, die über Mannose-6-Phosphat-Rezeptor abhängige Bahnen auf das Lysosom gerichtet sind, wird GCase vom ER durch den GCase-Transporter lysosomales integrales Membranprotein-2 (LIMP2) transportiert, das durch das Gen SCARB2 kodiert wird [6]. Einmal im Lysosom, interagiert das Enzym mit einem anderen Partner, seinem Aktivatorprotein Saposin C (SAPC) [7], einer Untereinheit des Vorläuferproteins Prosaposin (PSAP). Im lysosomalen Kompartiment hydrolysiert das Enzym Glucose-Einheiten sowohl aus GlcCer als auch aus Glucosylsphingosin (GlcSph) (Abb. 2).
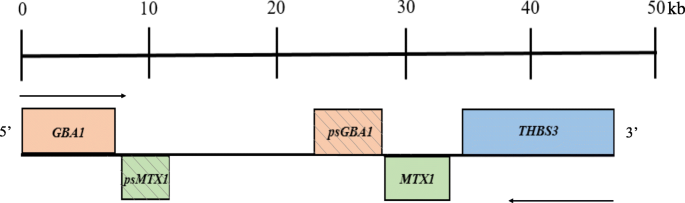
Das GBA1-Gen befindet sich in einer genreichen Region auf dem Chromosom 1q21, es besteht aus 11 Exons und umfasst etwa 7000 Basenpaare DNA [8]. Ein hochhomogenes, unübersetztes Pseudogen, das 98% Homologie in den kodierenden Regionen teilt, befindet sich nur 16 kb stromabwärts. Ein zweites Gen, Metaxin 1 (MTX1), das für ein Protein in der äußeren Mitochondrienmembran kodiert, liegt stromabwärts der GBA1-Pseudogensequenz und wird konvergent transkribiert [9]. Es gibt auch ein MTX1-Pseudogen, das sich zwischen GBA1 und seinem Pseudogen befindet. Das Gen für Thrombospondin 3 (TPS3), ein Glykoprotein, das Zell-zu-Matrix- und Zell-Zell-Interaktionen vermittelt, liegt unmittelbar stromabwärts von MTX1 (Abb. 3).
Mindestens 495 bekannte GBA1-Mutationen sind mit der Gaucher-Krankheit assoziiert, wobei es sich bei den meisten um Missense-Mutationen handelt [10, 11]. Die Nomenklatur der Mutation ist kompliziert, da die Nummerierung der mutierten Aminosäure vor einigen Jahren geändert wurde, um die 39-Aminosäure-Leader-Sequenz (neuere Nummerierung in Klammern) einzubeziehen. Es gibt zwei häufige Mutationen, die bei Patienten gefunden werden. Die Mutation N370S (p.N409S), die ausschließlich bei Patienten mit der Typ-1-Gaucher-Krankheit gefunden wurde, ist die häufigste Mutation, die bei Patienten in den Vereinigten Staaten, Europa und Israel gefunden wurde. Die Mutation L444P (p.L483P) wird weltweit gefunden und ist, wenn sie homozygot ist, häufig mit der neuronopathischen Gaucher-Krankheit assoziiert. Andere identifizierte Mutationen, die über alle Exons von GBA1 verteilt sind, umfassen Punktmutationen, Frame-Shifts, Spleißmutationen und Null-Allele, die häufig aus der Rekombination mit der homologen Pseudogensequenz resultieren [12]. Viele GBA1-Mutationen sind relativ häufig, aber andere sind seltener und nur in einzelnen Familien zu finden.
Quelle: https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-019-0336-2
Übersetzt mit www.DeepL.com/Translator