natur.de | O’Sullivan, Justin M. | Deutsche Übersetzung mit DeepL
Zusammenfassung
Die Erforschung der Parkinson-Krankheit (PD) hat sich weitgehend auf die Krankheit als eine einzelne Entität konzentriert, die sich auf die Entwicklung einer neuronalen Pathologie im zentralen Nervensystem konzentriert. Es setzt sich jedoch zunehmend die Erkenntnis durch, dass Morbus Parkinson keine Einzelerkrankung ist, sondern dass es sich um eine Mehrfacherkrankung handelt, bei der verschiedene Kombinationen von umweltbedingten, genetischen und potenziell komorbiden Faktoren zusammenwirken und den individuellen Krankheitsverlauf steuern. Darüber hinaus weisen immer mehr neuere Forschungsergebnisse darauf hin, dass periphere Gewebe und nicht-neuronale Zelltypen an der Entstehung von Parkinson beteiligt sind. Diese Beobachtungen stehen im Einklang mit der Hypothese, dass die ersten ursächlichen Veränderungen für die Entwicklung von Morbus Parkinson nicht im zentralen Nervensystem stattfinden müssen. Hier wird erörtert, wie die Verwendung der neuronalen Pathologie als gemeinsamer, qualitativer Phänotyp die Einblicke in die Möglichkeit multipler Ursprünge und Ätiologien von Morbus Parkinson minimiert. Darüber hinaus erörtern wir, wie die Betrachtung von Morbus Parkinson als eine einzige Entität unser Verständnis der ursächlichen molekularen Mechanismen, der Ansätze zur Patientenstratifizierung, der Identifizierung von Biomarkern und der Entwicklung von Therapieansätzen für Morbus Parkinson beeinträchtigen kann. Die eindeutige Konsequenz aus der Tatsache, dass es verschiedene Krankheiten gibt, die zusammen die Parkinson-Krankheit bilden, ist, dass es keinen einzigen Biomarker oder eine Behandlung für die Entwicklung oder das Fortschreiten der Krankheit gibt. Wir schlagen vor, die Diagnose von den klinischen Definitionen auf biologisch definierte Krankheiten zu verlagern, die zusammen die Parkinson-Krankheit bilden, um eine aussagekräftige Patientenstratifizierung zu ermöglichen. Klinische Designs vom Typ N-of-one bieten einen unvoreingenommenen und unabhängigen Ansatz zur Neudefinition von Morbus Parkinson als eine Gruppe von vielen einzelnen Krankheiten.
Einleitung
Es setzt sich zunehmend die Erkenntnis durch, dass die Parkinson-Krankheit keine einheitliche Erkrankung ist1,2. Vielmehr gibt es eine Vielzahl unterschiedlicher klinischer, genetischer und epidemiologisch heterogener Erkrankungen, die unter dem Oberbegriff der Parkinson-Krankheit zusammengefasst werden3,4,5. Der Einfachheit halber und um die Literatur nicht durch einen neuen Begriff zu verwirren, bezeichnen wir die verschiedenen Krankheiten im Folgenden als „Parkinson“. Trotz der zunehmenden Anerkennung dieses Konzepts konzentriert sich die Mehrzahl der auf Morbus Parkinson ausgerichteten Forschungsarbeiten auf den „gemeinsamen“ pathologischen Endpunkt einer linearen Morbus-Parkinson-Story6: die physische Manifestation neuronaler Einschlüsse, die so genannten Lewy-Körperchen, und den Verlust dopaminerger Neuronen (DAn) im zentralen Nervensystem (ZNS). Diese Konzentration auf den Endpunkt der Pathologie hat sich bei der Entwicklung wirksamer symptomatischer Therapien, zu denen auch Levodopa gehört, bewährt7. Das Scheitern von neunzehn Phase-3-Interventionsstudien8 , die auf eine Veränderung des Krankheitsverlaufs abzielten, verdeutlicht jedoch eine Einschränkung dieses Fokus. Der eingeschränkte Fokus auf den Endpunkt Pathologie ist u. a. darauf zurückzuführen, dass die Diagnose von Morbus Parkinson in der Regel erst viele Jahre nach dem Ausbruch der Krankheit gestellt wird, und zwar vorwiegend auf der Grundlage motorischer Symptome, und dass man Morbus Parkinson-Patienten erst nach der klinischen Diagnose untersuchen kann. Die erfolgreiche Entwicklung krankheitsmodifizierender Therapeutika wurde außerdem durch das Fehlen von Biomarkern und – was noch kritischer ist – durch das Fehlen aussagekräftiger molekularer Mechanismen behindert, die jede der einzelnen Krankheiten, die zusammen die Parkinson-Krankheit bilden, definieren. Dies spiegelt sich in einem Mangel an PD-Interventionsstudien wider, die auf spezifische mechanistische Veränderungen in Gruppen einzelner Patienten abzielen, die nach dem/den Mechanismus(en) definiert sind, die zur Entwicklung/Progression der Krankheit beitragen. Die SURE-PD3-Studie ist eine Ausnahme, die sich nur auf Personen mit niedrigen Serumuratkonzentrationen konzentrierte9. Abgesehen von der SURE-PD3-Studie gibt es jedoch in der Regel kein spezifisch messbares biologisches Signal für den Erfolg einer krankheitsmodifizierenden Intervention für jede einzelne Krankheit innerhalb des PD-Daches10. Stattdessen sind wir nach wie vor auf relativ unempfindliche und variable klinische Messgrößen für das Fortschreiten der Erkrankung angewiesen8.
Das Aufkommen genomweiter Assoziationsstudien (GWAS) hat die Identifizierung von Varianten ermöglicht, die mit dem Risiko der Krankheitsentwicklung11, unterschiedlichen Raten des kognitiven Verfalls12 und unterschiedlichen Raten des Fortschreitens von Parkinson in Verbindung gebracht werden13,14. Die Anhäufung von Datensätzen, die erforderlich ist, um die für GWAS erforderliche Stichprobengröße und statistische Aussagekraft zu erreichen, führt jedoch dazu, dass das Ein-Krankheits-Modell der Parkinson-Krankheit fortbesteht und das Vorhandensein mehrerer unterschiedlicher klinischer, genetischer und epidemiologisch heterogener Krankheiten übersehen wird. In diesen Situationen verwässert die Ansammlung von Daten über mehrere verschiedene Parkinson-Erkrankungen die Häufigkeit spezifischer krankheitsassoziierter Varianten und verringert somit die Fähigkeit, diejenigen Varianten zu identifizieren, die zum Verlauf jeder einzelnen Krankheit (d. h. zur Ätiologie) beitragen. Die Integration genetischer und standardisierter klinischer Daten in ein kohärentes, koordiniertes Konzept zur Verlangsamung oder Verhinderung der Entwicklung von Morbus Parkinson muss daher erst noch verwirklicht werden. Um dies zu erreichen, müssen wir uns von der Abhängigkeit von der gemeinsamen Endpathologie und den klinischen Definitionen lösen und ein Mittel zur Patientenstratifizierung unter Verwendung spezifischer genetischer Informationen entwickeln, das auf einem soliden Verständnis der Ätiologie jeder beitragenden molekularen Krankheit beruht. Aber wie kann man das erreichen, wenn man die verschiedenen Krankheiten erst definieren muss, um sie zu untersuchen? Im Folgenden wird erörtert, wie dieses zirkuläre Argument durch die Verwendung genetischer, molekularer und klinischer Informationen durchbrochen werden kann, um die verschiedenen Krankheitsverläufe innerhalb der Parkinson-Krankheit aus einer prospektiven, auf das Krankheitsrisiko ausgerichteten Perspektive zu identifizieren, die Patienten und Therapeutika ohne a priori-Annahmen stratifiziert.
Mehrere Krankheitsverläufe unter dem Dach der Parkinson-Krankheit
Im Jahr 2008 schrieb William Weiner: „Es gibt keine Parkinson-Krankheit „1 und schlug Parkinson-Krankheiten als passenderen Begriff für die beobachteten multiplen Ätiologien vor. Der Begriff Parkinson-Krankheit steht im Einklang mit der Tatsache, dass es nach der Diagnose keinen offensichtlichen, vorhersehbaren Krankheitsverlauf gibt, auch nicht bei monogenen Formen der Krankheit. Vielmehr ist der Krankheitsverlauf bei jedem Einzelnen einzigartig oder höchstens mit einer begrenzten Anzahl von Mitpatienten gemeinsam15.
Um zu veranschaulichen, welche Auswirkungen die Behandlung von Morbus Parkinson als eine Gruppe von Krankheiten mit unterschiedlichen, aber sich überschneidenden Ätiologien4,5,16 auf unser Verständnis der Krankheit haben kann, betrachten wir ein konzeptionelles Modell, in dem jede Krankheit innerhalb von Morbus Parkinson durch einen Berg innerhalb einer Reihe von Bergen dargestellt wird (Abb. 1). Da wir derzeit nicht in der Lage sind, die Anzahl der verschiedenen Krankheiten, die zusammen die Parkinson-Krankheit ausmachen, genau zu definieren, haben wir unser Modell der Einfachheit halber auf sieben Berge beschränkt. Im Morbus-Parkinson-Gebirgsmodell wird das genetische Risiko eines Individuums durch die Position im Tal (d. h. das Basislager) repräsentiert, an der das Individuum mit dem Aufstieg beginnt – diese Position schränkt natürlich die Berge, die bestiegen werden können, und die Route(n), die genommen werden können, ein. Morbus-Parkinson-Patienten gruppieren sich entsprechend ihres Basislagers, von denen es nur eine begrenzte Anzahl gibt und die durch die potenziellen und realisierten Kombinationen der Risikovarianten im Genom definiert sind. Umweltsignale aus der dynamischen Umgebung des Basislagers interagieren mit den individuellen genetischen Faktoren, um Aspekte der Krankheit zu verändern, einschließlich des Alters, in dem der Patient mit dem Klettern beginnt, oder ob die Person überhaupt Parkinson entwickelt. Zu diesen Umweltsignalen gehören u. a.: Pestizide und Schadstoffe17,18, Ernährung19, Virusinfektionen20, Kopftraumata21, Entzündungskrankheiten22 (für einen ausführlichen Überblick über die Rolle von Umweltsignalen im Zusammenhang mit der Genetik von Morbus Parkinson siehe Johnson et al.23). Sobald ein Individuum begonnen hat, einen Berg zu erklimmen, beeinflusst die Topologie des Berges, die die intrinsischen (z. B. das Darmmikrobiom24 oder komorbide Krankheitspathologie25) und extrinsischen (z. B. Bewegung26, Ernährung19 und regelmäßiges Fasten27) Faktoren darstellt, wie schnell jedes Individuum die Route erklimmt (d. h. die Geschwindigkeit der Krankheitsprogression) und somit die Präsentation und Schwere der Symptome15.
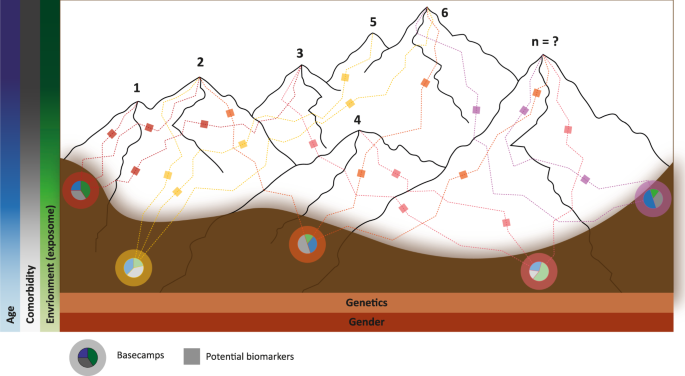
Konzeptuelles Modell, das die verschiedenen Krankheiten innerhalb der Parkinson-Krankheit mit Bergen innerhalb einer Gebirgskette gleichsetzt.
Es gibt wahrscheinlich viel mehr Berge (Krankheiten) als in diesem konzeptionellen Modell dargestellt. Die Topologie der Talsohle stellt die Gesamtvariation der Interaktion zwischen Alter, Umwelt, Komorbiditäten, Geschlecht und Genetik der Bevölkerung dar. Das genetische Risiko eines Individuums wird durch die Position im Tal (d. h. das Basislager) dargestellt, an der das Individuum mit dem Aufstieg beginnt. Verschiedene Signale (Umwelt, Alter, Komorbidität) aus der dynamischen Umgebung des Basislagers interagieren mit den genetischen Faktoren des Individuums, um Aspekte der Krankheit zu verändern, einschließlich des Alters, in dem der Patient mit dem Klettern beginnt, oder ob das Individuum überhaupt eine Parkinson-Erkrankung entwickelt (was sich in den Tortendiagrammen der Basislager widerspiegelt). Die Topologie des Berges (z. B. intrinsische und extrinsische Faktoren) wirkt sich darauf aus, wie schnell jeder Einzelne die Route erklimmt (d. h. die Geschwindigkeit des Krankheitsfortschritts), und damit auch auf die Bandbreite, das Auftreten und den Schweregrad der Symptome15. Die kleinen Kästchen (d. h. Kontrollpunkte) entlang der Aufstiegsrouten stellen potenzielle Biomarker dar, die entwickelt/genutzt werden könnten, um eine unvoreingenommene Momentaufnahme zu liefern, mit der sich die Krankheitsentwicklung bei einzelnen Patienten verfolgen lässt. Allerdings werden sich diese Biomarker im Verlauf der Krankheit wahrscheinlich verändern.
Die einzelnen Krankheiten, aus denen sich die Parkinson-Krankheit zusammensetzt, sind an und für sich heterogen. Dies wird in unserem Modell durch die Existenz mehrerer Routen zu jedem Berggipfel dargestellt. Diese Routen sind nicht unabhängig, sondern gehen ineinander über, was bedeutet, dass Individuen je nach ihrer speziellen Kombination von intrinsischen und extrinsischen Faktoren zwischen den Routen wechseln können. Obwohl jede Krankheit wahrscheinlich heterogen ist, wäre sie im Idealfall homogen genug, um ein einziges therapeutisches Ziel für die Entwicklung einer Behandlung zu bieten. Darüber hinaus gibt es für jeden Weg unterschiedliche Marker oder Kontrollpunkte in verschiedenen Stadien – ähnlich den Biomarkern, die eine unvoreingenommene Momentaufnahme liefern, mit der sich die Krankheitsentwicklung bei einzelnen Patienten verfolgen lässt. Es ist jedoch wichtig zu beachten, dass sich diese Biomarker im Verlauf der Krankheit verändern und wahrscheinlich durch das Alter des Patienten, seine Ernährung und die Kombination prädisponierender Begleiterkrankungen beeinflusst werden.
Es kann argumentiert werden, dass es Gemeinsamkeiten zwischen den einzelnen Krankheiten gibt, die zur Parkinson-Krankheit beitragen (d. h. zwischen den verschiedenen Bergen innerhalb des Spektrums). Durch die Behandlung dieser Gemeinsamkeiten könnte eine größere Gruppe von Patienten behandelt werden. Das mag richtig sein. Die Behandlung dieser Gemeinsamkeiten wäre zwar potenziell nützlich, hätte aber nur einen begrenzten Nutzen, da die Symptome (z. B. Ruhetremor und Bradykinesie) erst spät im Krankheitsverlauf auftreten und die Patienten daher zu dem Zeitpunkt, an dem die Behandlung eingeleitet wird, bereits stärker behindert sind (näher an ihren jeweiligen Gipfeln). Insbesondere haben krankheitsverändernde Maßnahmen, die auf dieser Grundlage auf Morbus Parkinson abzielen, bisher wenig Erfolg gehabt.
Andere Modelle von Parkinson wurden bereits vorgestellt. Am bekanntesten ist wohl das Elefantenmodell von William Langston28 , das die Idee einer vielfältigen Symptomatik aufgreift, Parkinson aber dennoch als eine einzige Krankheit, einen Elefanten, darstellt. In unserem Modell würde der Elefant als ein einzelner Berg innerhalb der Parkinson-Bergkette dargestellt. Langstons Modell erfasst also nicht die zahlreichen Krankheiten, die zusammen die Parkinson-Krankheit ausmachen, oder die Heterogenität, die jeder Krankheit eigen ist.
Omics zur Aufklärung der Entstehung und des Verlaufs der Parkinson-Krankheit nutzen
Es ist die Kombination von genetischen Risikokodierungen (z. B. LRRK2-G2019S oder SNCA-A53T) und nicht-kodierenden Varianten des Patienten, die zunächst „die Weichen stellen“ und bestimmen, welches Basislager und welchen Berg ein Individuum auf seiner Reise zur Parkinson-Krankheit besteigen wird. Die Anwendung von GWAS auf die Untersuchung von Morbus Parkinson ermöglicht eine unvoreingenommene Identifizierung der genetischen Risikogrundlagen auf Bevölkerungsebene, die lange vor Ausbruch der Krankheit bestehen. Die genetischen Varianten, die durch GWAS mit Morbus Parkinson in Verbindung gebracht wurden (z. B. 90 genetische Loci11), erklären jedoch nur zwischen 16 und 36 % der Erblichkeit von Morbus Parkinson. Darüber hinaus sind die Odds Ratios der einzelnen Varianten, von wenigen Ausnahmen abgesehen, in der Regel niedrig (z. B. zwischen 0,8 und 1,2)11. Tatsächlich ist die derzeitige Vorhersagefähigkeit der mit Morbus Parkinson assoziierten SNPs so gering, dass eine aussagekräftige Risikoscore-Prognose nicht durchführbar ist29. Die fehlende Heritabilität lässt sich zum Teil durch Probleme bei der Zusammenführung der vielen verschiedenen Krankheiten, die zu Morbus Parkinson beitragen, zu einer einzigen Einheit erklären, die durch pathologische Marker im Spätstadium definiert wird (d. h. die Durchführung von GWAS aus der Perspektive, dass Morbus Parkinson eine einzige Krankheit ist). Darüber hinaus bedeutet die Abhängigkeit von einer klinischen Definition, dass keine zwei „Omics“-Studien ähnliche Ergebnisse liefern, da sie nur die Ergebnisse der heterogenen Patientenpopulation repräsentieren, an der sie durchgeführt wurden (z. B. 30). Die Mittelung dieser unterschiedlichen, aber verwandten Datensätze führt dazu, dass nur die signifikantesten Risikoloci identifiziert werden, die für alle Krankheiten gleich sind und statistische Signifikanz erreichen. Das Problem der Mittelung von Signalen über die heterogenen Krankheiten, die zu Morbus Parkinson beitragen, kann bei der Durchführung einer GWAS dadurch gelöst werden, dass Morbus Parkinson-Patienten nach ihrem genetisch definierten Ausgangspunkt stratifiziert werden, was wiederum die Auswahl informativer Längsschnitt-Biomarker und wirksamer therapeutischer Ansätze (spezifisch für jeden Weg) ermöglicht. Diese Stratifizierung kann durch genomische Ansätze erreicht werden, die die Besonderheiten der GWAS-Manifestation31,32 erforschen und über die verschiedenen Entwicklungswege von Parkinson informieren. So könnte eine GWAS-basierte Patientenstratifizierung Aufschluss darüber geben, 1) welche(r) Signalweg(e) gestört ist/sind, 2) welche Signalweg-Biomarker untersucht werden sollten und 3) welche Ziele für therapeutische Maßnahmen in Betracht gezogen werden sollten. Der Übergang von der einfachen Identifizierung von GWAS-Signalen zur informativen Stratifizierung erfordert jedoch eine eingehende Charakterisierung der Funktion der ursächlichen Variante(n)33.
Bis vor kurzem33 waren wir nicht in der Lage, nicht-kodierende genetische Variationen und Risiken funktionell in biologisch krankheitsrelevante Pfade zu übersetzen, was bedeutete, dass die früheren Stadien der Parkinson-Entwicklung als Mittel zur Stratifizierung oder therapeutischen Intervention hauptsächlich vernachlässigt wurden. Im Gegensatz zu den nicht-kodierenden Risikovarianten wurden kodierende Mutationen in den GBA- und LRRK2-Genen erforscht und ermöglichten eine Stratifizierung von Patienten mit diesen spezifischen Mutationen für therapeutische Interventionen, die auf diese genetischen Untergruppen von Patienten ausgerichtet sind34,35. Darüber hinaus wiesen Szwedo et al. nach, dass APOE-ε4- und GBA-Mutationen eine Rolle für die Geschwindigkeit des kognitiven Verfalls bei Morbus Parkinson-Patienten spielen, fanden aber keine signifikanten Auswirkungen für häufige Varianten in SNCA und MAPT12. Diese Ergebnisse eröffnen die Möglichkeit, Personen mit einem hohen Risiko für einen raschen kognitiven Verfall früher zu identifizieren und zu stratifizieren und so geeignete Kandidaten für künftige gezielte Studien zu identifizieren. Trotz des Fortschritts ist die bekannte unvollständige Penetranz dieser Mutationen problematisch36 und weist auf eine verbleibende Wissenslücke in Bezug auf die mechanistische Rolle einiger dieser Mutationen hin, wie z. B. die Rolle von LRRK2-Mutationen bei der Krankheitsprogression14. Es stellt sich daher die Frage, ob solche Maßnahmen auch bei Patienten mit diesen spezifischen Mutationen das Fortschreiten der Krankheit aufhalten können. Nichtsdestotrotz entwickelt sich mit den jüngsten Fortschritten unser Verständnis dafür, wie sich sowohl kodierende als auch nicht-kodierende Risiken manifestieren33,37. Dieses Verständnis kann zur Aufstellung von Hypothesen genutzt werden, die zur Identifizierung und strengeren Klassifizierung einzelner Krankheiten innerhalb der Parkinson-Krankheit beitragen, was auch zu gezielten Therapien führen könnte.
Die funktionelle Charakterisierung setzt voraus, dass die damit verbundene molekulare, zelluläre und physiologische Phänotypisierung ausreichend tiefgreifend ist, um eine genaue Zuordnung der ursächlichen Varianten und ihrer Zielgene zu ermöglichen38 und möglicherweise zu bestimmen, in welchen Geweben sich das Krankheitsrisiko manifestiert. Panyard et al. wendeten einen Ansatz an, um die Wirkung kausaler genetischer Varianten bei der Alzheimer-Krankheit (AD)39 funktionell zu charakterisieren und zuzuordnen. Panyard et al. integrierten genomische und klinische Daten aus zwei Längsschnittkohorten der Alzheimer-Krankheit mit epigenetischen Annotationen, um zelltypspezifische genomische Funktionsannotationen zu entwickeln39. Diese Anmerkungen wurden verwendet, um zu ermitteln, welche SNPs in verschiedenen Geweben wahrscheinlich funktionell sind39. Die Autoren wiesen nach, dass die Auswirkungen dieser SNPs in der Leber statistisch mit der Alzheimer-Diagnose verbunden sind39. Damit wiesen Panyard et al. auf einen potenziellen Beitrag der Leber zur Alzheimer-Krankheit hin, einschließlich Assoziationen mit zentralen Biomarkern der Alzheimer-Krankheit im Liquor, die weithin als „hirnzentrierte“ Krankheit angesehen wird. Es handelt sich zwar um eine kleine Studie (n = 79 Alzheimer-Patienten), aber die Feststellung, dass Veränderungen in der Leber für einige, aber nicht für alle Personen prädiktiv waren, steht im Einklang mit der Hypothese, dass die Fehlfunktion der Leber für eine der heterogenen Krankheiten verantwortlich ist, die gemeinsam zu Alzheimer beitragen40.
Genomische Ansätze werden auch bei Versuchen angewandt, die Zell- und Gewebetypen zu identifizieren und zu verstehen, in denen sich das genetische Risiko bei Parkinson manifestiert41,42. Coetzee et al.43 beispielsweise nutzten Daten über Histonveränderungen in Kombination mit Anreicherungsanalysen, um zu zeigen, dass viele mit Morbus Parkinson assoziierte genetische Varianten in nicht-neuronalen Zelltypen, einschließlich Lymphozyten, Mesendoderm-, Leber- und Adipozytenzellen, angereichert sind und mit quantitativen Expressionsmerkmalen (eQTL) assoziiert sind43. In ähnlicher Weise haben wir einen entdeckungsbasierten Ansatz verwendet, um mutmaßliche regulatorische Auswirkungen von nicht-kodierenden PD-assoziierten Risikovarianten sowohl im ZNS als auch in peripheren Geweben zu identifizieren33,44. Unsere Analysen ergaben, dass eQTL-Effekte für eine Untergruppe (28 %) der 90 PD-assoziierten Risikovarianten nur in peripheren Geweben (z. B. Schilddrüse und Speiseröhre) nachgewiesen wurden33 , während nur 2 % der PD-Risiko-SNPs ausschließlich in ZNS-Geweben identifizierbare eQTLs aufwiesen. Da es sich bei den Geweben um eine komplexe Mischung von Zelltypen handelt, bedeutet der Befund in der Speiseröhre nicht, dass der Effekt auf die Muskeln zurückzuführen ist und nicht auf die Nerven, die die Speiseröhre versorgen. Der Befund steht jedoch im Einklang mit peripheren Symptomen (z. B. Dysphagie), die manchmal im Frühstadium von PD45 beobachtet werden.
In einem Versuch, festzustellen, welche Gewebe und damit Zelltypen für die Erblichkeit von Morbus Parkinson verantwortlich sind, verwendeten Reynolds et al.41 eine stratifizierte Linkage-Disequilibrium-Score-Regression46 (siehe Kasten 1), um den Beitrag gemeinsamer genetischer Variationen zur Erblichkeit von Morbus Parkinson in 53 Geweben (einschließlich 13 Hirnregionen) zu messen, wobei Schizophrenie als Vergleichsmaßstab diente. Im Gegensatz zur Schizophrenie, bei der alle 13 Hirngewebe signifikant für die Heritabilität angereichert waren, gab es in keinem der 53 Gewebe (im ZNS oder in peripheren Geweben) eine Anreicherung für die PD-Erblichkeit. Die fehlende Anreicherung der PD-Heritabilität in diesen Massengeweben veranlasste Reynolds et al. zu der Frage, ob die zelluläre Heterogenität innerhalb der Gewebe möglicherweise Signale maskiert, und so versuchten sie, die zelltypspezifische Anreicherung der Heritabilität zu untersuchen. Reynolds et al. konnten jedoch bei 6 Zelltypen des menschlichen und 30 ZNS-Zelltypen der Maus keine zelltypspezifische Anreicherung der PD-Erblichkeit feststellen. Die Lewy-Body-Pathologie in bestimmten neuronalen Zelltypen, die mit Morbus Parkinson in Verbindung gebracht wird, hat die Forscher dazu veranlasst, ihre Bemühungen auf das Verständnis des Risikos in neuronalen Subtypen zu konzentrieren. Die Ergebnisse von Reynolds et al. geben jedoch Grund zu der Annahme, dass die Risikoloci auch andere Zelltypen als das ZNS und/oder zelluläre Prozesse und Wege über mehrere Zelltypen hinweg betreffen und für die die verschiedenen Zelltypen unterschiedlich anfällig sind41. Eine solche unterschiedliche Anfälligkeit, die mit der vorgeschlagenen Schwellentheorie für PD47 übereinstimmt, könnte wahrscheinlich das Ergebnis von Wechselwirkungen mit Umweltfaktoren und/oder komorbider Krankheitspathologie sein.
Im Gegensatz zu der von Reynolds et al. festgestellten fehlenden Anreicherung der Erblichkeit von Zelltypen gibt es bisher mehrere Studien, die gliale Zelltypen, vor allem Mikroglia, in die Neuroinflammation und die Pathogenese von Parkinson einbeziehen42,48,49. Angesichts dieser Zusammenhänge kombinierten Bryois et al. zelltypspezifische Genexpressions- und GWAS-Daten, um die Rolle der Gliazellen bei der Entstehung von Parkinson zu untersuchen49. Ein Hinweis auf die Rolle der Mikroglia war die Feststellung, dass zelltypspezifische ATACseq-Daten funktionelle PD-Risikoloci identifizierten, die für Autophagie und lysosomale Prozesse angereichert waren50, die beide schon früher mit PD in Verbindung gebracht wurden51. Darüber hinaus hat sich gezeigt, dass eine erhöhte LRRK2-Expression, die mit den PD-GWAS-SNPs rs76904798 und rs7294619 (R2 = 0,842) verbunden ist, speziell in Mikroglia auftritt42. Insgesamt stimmen diese Daten mit der Hypothese überein, dass genetische Risikovarianten für Morbus Parkinson nicht-neuronale Zelltypen des ZNS betreffen. Auch wenn diese Studien die Bedeutung der Berücksichtigung von Zelltypen unterstreichen, beruhen sie dennoch auf a priori-Annahmen, die sich auf das ZNS konzentrieren. Daher ist es von entscheidender Bedeutung, diese Analysen auf Nicht-ZNS-Zelltypen auszudehnen und dabei einen eher entdeckungsbasierten, hypothesenfreien Ansatz zu verfolgen, um festzustellen, ob eine solche Risikoanreicherung wirklich spezifisch für die Mikroglia ist oder ob auch andere Nicht-ZNS-Zelltypen an der Krankheitsentstehung und -ausbreitung beteiligt sein könnten.
Zusammengenommen zeigen diese Studien, wie mehrere „omics“-Ansätze verwendet werden können, um die gewebe- und zelltypspezifischen Manifestationen für GWAS-Risikovarianten zu identifizieren. Die von uns diskutierten Ergebnisse unterstützen zwei mögliche, sich nicht gegenseitig ausschließende Hypothesen: Erstens können die einzelnen Krankheiten innerhalb der Parkinson-Krankheit durch genetische Variationen abhängige Mechanismen entstehen, die das/die Ursprungsgewebe und damit die mit der Krankheit verbundenen pathologischen Pfade bestimmen. Dieses Konzept spiegelt sich in dem Modell der Gebirgskette wider, wobei jedes Basislager einen anderen, genetisch bedingten Ausgangspunkt darstellt. Die zweite Hypothese besagt, dass Varianten, die sich auf einen bestimmten peripheren Gewebe- oder Zelltyp auswirken, eine Dysregulation verursachen, die die Komplexität/Symptome der Krankheit verstärkt, ohne notwendigerweise zu der ZNS-Pathologie zu führen, die typischerweise mit Morbus Parkinson in Verbindung gebracht wird. Diese zweite Hypothese stimmt mit der Schwellentheorie für Morbus Parkinson überein, die auf der Grundlage einer parallelen Degeneration sowohl des zentralen als auch des peripheren Nervensystems entwickelt wurde47. Es ist also notwendig, über die Gewebe- und Zelltypen hinauszublicken, die traditionell mit der Parkinson-Pathologie in Verbindung gebracht werden, um ein besseres Verständnis der Mechanismen zu erlangen, durch die sich das genetische Risiko manifestieren kann. Fortschritte auf dem Gebiet der Einzelzell-Transkriptomik52,53 und Bulk-Zell-Analysen54 werden zusätzliche Erkenntnisse liefern, die die relativen Beiträge der Genetik und der Umwelt zur Manifestation des Parkinson-Risikos zu entwirren beginnen. Es stellt sich jedoch die Frage, wie wir diese Ansätze auf eine mechanistisch-heterogene Krankheit anwenden können.
Nutzung von Big Data zur Identifizierung individueller Verläufe bei einer heterogenen Krankheit
Die Zusammenführung von Daten aus verschiedenen Kohorten ermöglicht eine große Stichprobengröße (n), die sonst mit einer einzigen Kohorte nicht erreicht werden kann. Somit bieten konglomerierte Daten die dringend benötigte statistische Aussagekraft, um bestimmte Hypothesen zu untersuchen. Trotz der statistischen Aussagekraft verdeutlicht die Zusammenfassung verschiedener Morbus-Parkinson-Kohorten leider auch das Fehlen strikter Diagnosekriterien für Morbus Parkinson und verwandte Krankheiten, da verschiedene Kohorten oft unterschiedliche Diagnosekriterien verwenden30. Ein weiteres Problem, das die Diagnose selbst auf der Ebene eines einzelnen Klinikers beeinträchtigt, ist die Fehlklassifizierung55. Eine solche Fehlklassifizierung wirft das Problem der Einbeziehung von Nicht-PD-Patienten in Kohorten auf, wodurch die Ergebnisse von Beobachtungsstudien und klinischen Versuchen verzerrt werden können. Ein dritter und wesentlicher erschwerender Faktor ist die Tatsache, dass in den derzeit untersuchten „homogenen“ klinischen Parkinson-Kohorten wahrscheinlich mehrere unterschiedliche mechanistische Erkrankungen vorliegen. Dieses Problem tritt besonders bei Kohorten auf, die Patienten mit unterschiedlichen genetischen Prädispositionen für Krankheiten innerhalb der Parkinson-Krankheit umfassen, wie z. B. GBA-PD- und LRRK2-PD-Patienten, die typischerweise einen unterschiedlichen symptomatischen Verlauf aufweisen56,57. Die Gruppierung dieser verschiedenen individuellen Krankheiten führt wahrscheinlich zu einem Informationsverlust. Wenn die Zusammenführung von Daten den erhofften Erfolg bringen soll, werden dringend Krankheits-Biomarker benötigt, und zwar insbesondere Biomarker für die verschiedenen Krankheiten, die zusammen die Parkinson-Krankheit bilden. Die Notwendigkeit, einzelne Krankheiten zu definieren, anstatt sie zu einer einzigen Einheit zu verschmelzen, steht im Einklang mit der Vorhersage von Espay und Lang, dass kleinere, intelligentere klinische Studien erforderlich sind, um von diesem „homogenen“ klinischen Parkinson-Phänotyp wegzukommen6.
Wie bereits erörtert, bieten genetische Risikovarianten eine Möglichkeit für eine solche genetische Stratifizierung – wobei das Risikoprofil einer Person den Ausgangspunkt ihrer Krankheit bestimmt (z. B. ein bestimmtes Basislager im Bergkettenmodell). Diese genetischen Risikovarianten (SNPs) wirken jedoch nicht unabhängig voneinander33. Vielmehr wirken sie auf kombinatorische Weise innerhalb eines viel größeren genetischen Hintergrunds. Um den vollen Beitrag der mit der Parkinson-Krankheit assoziierten SNPs zur Parkinson-Krankheit zu verstehen, müssen sie im Kontext des omnigenischen58 und infinitesimalen59 Krankheitsmodells (siehe Kasten 1) und im Sinne der Netzwerkmedizin60 betrachtet werden. Mit netzwerkmedizinischen Ansätzen kann die Krankheit als eine Summe miteinander verbundener Störungen kontextualisiert werden, die die zugrunde liegenden genetischen und molekularen Risikofaktoren widerspiegeln (d. h. die Untersuchung von PD-Risikovarianten im Kontext des gesamten Genotyps einer Person). Der Nutzen der Netzwerkmedizin60 wurde bereits bei anderen komplexen Krankheiten erforscht und hat bereits zur Identifizierung neuer Ziele für therapeutische Strategien und deren Entwicklung beigetragen61,62.
Die Erforschung der Auswirkungen und der Interkonnektivität der genetischen Beiträge zum individuellen Krankheitsrisikoprofil unter dem Gesichtspunkt der Netzwerkmedizin ist erst nach den jüngsten Fortschritten möglich geworden. Dazu gehören die Senkung der Kosten für die Genomsequenzierung und die Datenverarbeitung63 sowie die Entwicklung von Ansätzen des maschinellen Lernens zur Erkennung komplexer Muster in Genomen. Diese Fortschritte haben die sich rasch entwickelnde Post-GWAS-Genom-Editierungs-Toolbox, einschließlich CRISPR-Screens64 und massiv-parallele Reporter-Assays65 (um beobachtete Muster auf ihre funktionelle Bedeutung hin zu testen), beeinflusst und wurden durch sie verstärkt. Diese Instrumente werden im Laufe der Zeit die Daten liefern, die erforderlich sind, um die vollständigen genetischen Beiträge zur Entwicklung der Krankheiten zu verstehen, die zusammengenommen die Parkinson-Krankheit und andere komplexe Krankheiten bilden. Gemeinsame Anstrengungen wie die Atlas of Variant Effects Alliance (https://www.varianteffect.org/)66 werden entscheidend dazu beitragen, die Ergebnisse dieser funktionellen Post-GWAS-Studien zu kuratieren und systematisch zusammenzustellen. Ein weiterer aktueller technologischer Fortschritt, der die genomischen Ergebnisse aus phänotypischer Sicht wahrscheinlich verbessern wird, ist die Einführung von tragbaren Geräten67. Es hat sich gezeigt, dass solche Geräte Vitaldaten (z. B. Herzfrequenz und elektrodermale Aktivität) auf einem Niveau liefern können, das dem in einer klinischen Umgebung gewonnenen entspricht68. Die weite Verbreitung dieser Wearables ermöglicht eine individualisierte, longitudinale und kontinuierliche Gesundheitsüberwachung. Während es eine Herausforderung ist, bei Bewegungsmessungen das Signal vom Rauschen zu unterscheiden, verspricht die Kombination der detaillierten phänotypischen Daten, die Wearables liefern, mit abgeglichenen genetischen Daten, bei der Identifizierung klinischer Unterschiede zwischen den verschiedenen genomischen Krankheiten innerhalb von Parkinson zu helfen.
Informationen über genetische Variationen und Medikamentenreaktionen können genutzt werden, um zu bestimmen, welche Medikamente bei einer Person wahrscheinlich sicher und wirksam sind. Diese Ansätze führen zur Entstehung von ‚genetisch informierten‘ klinischen Studien (d. h. präzisionsmedizinischen Ansätzen) bei Parkinson69,70,71. So wurde beispielsweise Ambroxol für die Behandlung von Parkinson-Patienten mit einer GBA-Kodierungsmutation neu ausgerichtet34. Obwohl Ambroxol nur in einer kleinen, offenen, nicht randomisierten Gruppe von Patienten erprobt wurde, ist es vielversprechend für die Behandlung dieser gut definierten, aber heterogenen (d. h. mehrere GBA-kodierende Mutationen enthaltenden) Untergruppe von Patienten34. Die Ambroxol-Studie ist ein Beispiel, das den Weg für künftige präzisionsorientierte klinische Studien bei Morbus Parkinson ebnet. Sie befasst sich nicht nur mit der Frage der Behandlung von Patienten auf der Grundlage genomischer Informationen, sondern zeigt auch das Potenzial der Umwidmung bereits zugelassener Medikamente72 , um den Prozess der Arzneimittelentwicklung zu beschleunigen. Die Ambroxol-Studie umfasste auch einige idiopathische Morbus-Parkinson-Patienten, die ebenfalls vielversprechende Reaktionen auf die Behandlung zeigten. Die Identifizierung von idiopathischen Morbus-Parkinson-Patienten, die spezifisch eine reduzierte GCase-Aktivität aufweisen (d. h. solche mit GBA-modifizierenden Genotypen30,44), könnte zu besseren Ergebnissen für die Patienten führen.
Trotz des offensichtlichen Versprechens eines stratifizierten Ansatzes für klinische Tests und Therapien bedeutet das Fehlen einer Genotypisierung als Teil der klinischen Beurteilung, dass die Identifizierung der relativ kleinen Anzahl von Personen mit genetischen Prädispositionen eine große finanzielle und zeitliche Herausforderung bleibt. Dies ändert sich jedoch mit Initiativen wie PD Frontline (https://pdfrontline.com/en) und PD GENEration (https://www.parkinson.org/PDGENEration), die Gentests für Parkinson-Patienten anbieten, um sicherzustellen, dass Personen, die bestimmte Mutationen tragen, an die für sie am besten geeigneten klinischen Studien verwiesen werden.
Schlussbemerkungen und Zukunftsperspektiven
Die Erkenntnis, dass viele Krankheiten zu Morbus Parkinson beitragen, macht deutlich, dass die Suche nach einem Biomarker für das Fortschreiten von Morbus Parkinson und nach Therapeutika eine Herausforderung darstellt. Wenn es viele Krankheiten gibt, die unter dem Begriff Morbus Parkinson zusammengefasst werden, dann sollten wir nach Biomarkern für jede einzelne Krankheit suchen. Die Tatsache, dass wir Patienten weiterhin nach klinischen und nicht nach biologischen Kriterien auswählen, beeinträchtigt unsere Fähigkeit, dies zu tun. Selbst das genetische Risiko für Morbus Parkinson wird derzeit im Zusammenhang mit der gemeinsamen Pathologie gesehen, die die verschiedenen Parkinson-Krankheiten miteinander verbindet. Der Nutzen der Netzwerkmedizin60 hat sich bei anderen komplexen Krankheiten bewährt und hilft bei der Identifizierung neuer Ziele für therapeutische Strategien und deren Entwicklung61,62. Es stimmt zwar, dass weitere Initiativen, die eine groß angelegte Datenzusammenführung beinhalten, zum molekularen und klinischen Verständnis der Krankheit beitragen werden, aber die mangelnde Einheitlichkeit der Parkinson-Diagnose und des Krankheitsverlaufs wird wahrscheinlich die Ergebnisse von Genom- und Biomarker-Studien beeinträchtigen30,73. Jüngste Initiativen (z. B. PREDICT-PD74 und das Cincinnati Cohort Biomarker Program (CCBP)75), die auf Entdeckungen basierende Analysen prospektiver Kohorten einbeziehen, versuchen, dieses Problem durch die Definition von PD-Entwicklungspfaden und Biomarkern zu lösen. Darüber hinaus sind wir der Meinung, dass es an der Zeit ist, in der Parkinson-Forschung systematische n-of-176,77,78-Ansätze (siehe Kasten 1) in Betracht zu ziehen, um die Kombinationen und relativen Beiträge der genetischen, pathologischen und umweltbedingten Faktoren in jedem einzelnen Fall3,79 für Individuen innerhalb einer heterogenen Population zu ermitteln. Die Nutzung von Wearables durch die Bevölkerung wird dazu beitragen, relevante Daten für einen solchen Ansatz zu sammeln80. Letztendlich werden die aggregierten Ergebnisse von n-of-1-Ansätzen dazu beitragen, die vielen Krankheiten aufzuklären, die zu der einen komplexen Parkinson-Krankheit beitragen. Die Neudefinition der Hypothesen, die die Parkinson-Forschung vorantreiben, wird eine Abkehr von der derzeitigen Konzentration auf eine gemeinsame Pathologie und klinische Definitionen ermöglichen. Dies wiederum wird den Weg frei machen für die Entwicklung gezielter diagnostischer und therapeutischer Ansätze, die auf einem molekularen Verständnis der Ätiologie der einzelnen Krankheiten beruhen und somit in der Lage sind, das Fortschreiten der Krankheit zu verlangsamen, zu stoppen oder umzukehren und letztlich die Krankheit zu verhindern.
Zusammenfassung der Berichterstattung
Weitere Informationen zum Forschungsdesign finden Sie in der Nature Research Reporting Summary, die mit diesem Artikel verlinkt ist.
Verfügbarkeit von Daten
Es gibt keine Daten, die sich speziell auf dieses Perspektivpapier beziehen. Alle Informationen sind in den Referenzen enthalten.
Literaturhinweise
Weiner, W. J. Es gibt keine Parkinson-Krankheit. Arch. Neurol. 65, 705-708 (2008).
Artikel PubMed Google Scholar
Espay, A. J. et al. Krankheitsveränderung und Entwicklung von Biomarkern bei der Parkinson-Krankheit. Neurologie 94, 481-494 (2020).
Artikel PubMed PubMed Central Google Scholar
Mestre, T. A. et al. Parkinson's Disease Subtypes: Critical Appraisal and Recommendations. J. Parkinsons. Dis. 11, 395-404 (2021).
Artikel PubMed PubMed Central Google Scholar
Espay, A. J., Brundin, P. & Lang, A. E. Precision medicine for disease modification in Parkinson disease. Nat. Rev. Neurol. 13, 119-126 (2017).
Artikel PubMed Google Scholar
Bloem, B. R., Okun, M. S. & Klein, C. Parkinson's Disease. Lancet 397, 2284-2303 (2021).
CAS Artikel PubMed Google Scholar
Espay, A. J. & Lang, A. E. Parkinson diseases in the 2020s and beyond: Ersetzen der klinisch-pathologischen Konvergenz durch systembiologische Divergenz. J. Parkinsons. Dis. 8, S59-S64 (2018).
Artikel PubMed PubMed Central Google Scholar
Paoletti, F. P., Tambasco, N. & Parnetti, L. Levodopa treatment in Parkinson's disease: early or later? Ann. Transl. Med. 7, S189-S189 (2019).
Artikel Google Scholar
Espay, A. J. & Stecher, B. Brain Fables: Die verborgene Geschichte der neurodegenerativen Krankheiten und ein Plan zu ihrer Überwindung. https://doi.org/10.1017/9781108888202 (Cambridge University Press, 2020).
Schwarzschild, M. A. et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 326, 926-939 (2021).
CAS Artikel PubMed Google Scholar
Espay, A. J. et al. Biomarker-driven phenotyping in Parkinson's disease: Ein fehlendes Glied in klinischen Studien zur Krankheitsmodifikation. Mov. Disord. 32, 319-324 (2017).
Artikel PubMed PubMed Central Google Scholar
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091-1102 (2019).
CAS Artikel PubMed PubMed Central Google Scholar
Szwedo, A. A. et al. GBA und APOE Impact Cognitive Decline in Parkinson's Disease: A 10-Year Population-Based Study. Mov. Disord. https://doi.org/10.1002/MDS.28932 (2022).
Tan, M. M. X. et al. Genome-Wide Association Studies of Cognitive and Motor Progression in Parkinson's Disease. Mov. Disord. 36, 424-433 (2020).
Artikel CAS PubMed Google Scholar
Liu, G. et al. Genomweite Überlebensstudie identifiziert einen neuartigen synaptischen Locus und polygenen Score für die kognitive Progression bei der Parkinson-Krankheit. Nat. Genet. 53, 787-793 (2021).
CAS Artikel PubMed PubMed Central Google Scholar
Ravan, A. et al. Nicht-motorische Symptome in einer indischen Kohorte von Parkinson-Patienten und Korrelation des Fortschreitens der nicht-motorischen Symptome mit der motorischen Verschlechterung. Neurol. India 63, 166 (2015).
Artikel PubMed Google Scholar
Berg, D. et al. Prodromale Subtypen der Parkinson-Krankheit - Schlüssel zum Verständnis der Heterogenität. Nat. Rev. Neurol. 17, 349-361 (2021).
Artikel PubMed Google Scholar
Pezzoli, G. & Cereda, E. Exposition gegenüber Pestiziden oder Lösungsmitteln und das Risiko einer Parkinson-Erkrankung. Neurology 80, 2035-2041 (2013).
CAS Artikel PubMed Google Scholar
Kamel, F. Paths from pesticides to Parkinson's. Science 341, 722-723 (2013).
CAS-Artikel PubMed Google Scholar
Paknahad, Z., Sheklabadi, E., Derakhshan, Y., Bagherniya, M. & Chitsaz, A. The effect of the Mediterranean diet on cognitive function in patients with Parkinson's disease: Eine randomisierte klinisch kontrollierte Studie. Complement. Ther. Med. 50, 102366 (2020).
Artikel PubMed Google Scholar
Olsen, L. K., Dowd, E. & McKernan, D. P. A role for viral infections in Parkinson's etiology? Neuronal Signal 2, 20170166 (2018).
Artikel Google Scholar
Paul, K. C. et al. The association between lifestyle factors and Parkinson's disease progression and mortality. Mov. Disord. 34, 58-66 (2019).
Artikel PubMed PubMed Central Google Scholar
Villumsen, M., Aznar, S., Pakkenberg, B., Jess, T. & Brudek, T. Inflammatory bowel disease increases the risk of Parkinson's disease: a Danish nationwide cohort study 1977-2014. Gut 68, 18-24 (2019).
CAS Artikel PubMed Google Scholar
Johnson, M. E., Stecher, B., Labrie, V., Brundin, L. & Brundin, P. Triggers, Facilitators, and Aggravators: Neudefinition der Pathogenese der Parkinson-Krankheit. Trends Neurosci. 42, 4-13 (2019).
CAS Artikel PubMed Google Scholar
Heinzel, S. et al. Gut Microbiome Signatures of Risk and Prodromal Markers of Parkinson Disease. Ann. Neurol. 88, 320-331 (2020).
Artikel PubMed Google Scholar
Cheong, J. L. Y., de Pablo-Fernandez, E., Foltynie, T. & Noyce, A. J. The Association Between Type 2 Diabetes Mellitus and Parkinson's Disease. J. Parkinsons. Dis. 10, 775-789 (2020).
CAS Artikel PubMed PubMed Central Google Scholar
Crotty, G. F. & Schwarzschild, M. A. Chasing Protection in Parkinson's Disease: Does Exercise Reduce Risk and Progression? Front. Aging Neurosci. 12, 1-11 (2020).
Artikel Google Scholar
Neth, B. J., Bauer, B. A., Benarroch, E. E. & Savica, R. The Role of Intermittent Fasting in Parkinson's Disease. Front. Neurol. 12, 1-7 (2021).
Artikel Google Scholar
Langston, J. W. Der Parkinson-Komplex: Parkinsonismus ist nur die Spitze des Eisbergs. Ann. Neurol. 59, 591-596 (2006).
Artikel PubMed Google Scholar
Koch, S. et al. Validität und prognostischer Wert eines polygenen Risikoscores für die Parkinson-Krankheit. Genes (Basel) 12, 1859 (2021).
CAS Artikel Google Scholar
O'Sullivan, J. M., Heijer, J. M., Groeneveld, G. J. & Cooper, A. A. Intronic Haplotypes in GBA Modify Age at Diagnosis of Parkinson's: Replikation in einer Untergruppe. Mov. Disord. 36, 1468-1470 (2021).
Artikel PubMed Google Scholar
Novikova, G. et al. Integration der Genetik der Alzheimer-Krankheit und der myeloischen Genomik identifiziert regulatorische Elemente und Gene des Krankheitsrisikos. Nat. Commun. 12, 1-14 (2021).
Artikel CAS Google Scholar
Wang, Q. et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson's disease. Nat. Commun. 10, 1-15 (2019).
Artikel CAS Google Scholar
Farrow, S. L. et al. Establishing gene regulatory networks from Parkinson's disease risk loci. Brain 139, 1-36 (2022).
Google Scholar
Mullin, S. et al. Ambroxol zur Behandlung von Patienten mit Parkinson-Krankheit mit und ohne Glukozerebrosidase-Genmutationen. JAMA Neurol. 77, 427-434 (2020).
Artikel PubMed PubMed Central Google Scholar
Ding, X. & Ren, F. Leucine-rich repeat kinase 2 inhibitors: a patent review (2014-present). Expert Opin. Ther. Pat. 30, 275-286 (2020).
CAS Artikel PubMed Google Scholar
Anheim, M. et al. Penetranz der Parkinson-Krankheit bei Trägern einer Glukozerebrosidase-Genmutation. Neurology 78, 417-420 (2012).
CAS Artikel PubMed Google Scholar
Ho, D. et al. Machine Learning Identifies Six Genetic Variants and Alterations in the Heart Atrial Appendage as Key Contributors to PD Risk Predictivity. Front. Genet. 12, 2409 (2022).
Artikel Google Scholar
Karthik Jagadeesh, A. A. et al. Identifizierung krankheitskritischer Zelltypen und zellulärer Prozesse im menschlichen Körper durch Integration von Einzelzellprofilen und Humangenetik. bioRxiv 2021.03.19.436212 https://doi.org/10.1101/2021.03.19.436212 (2021).
Panyard, D. J. et al. Liver-specific polygenic risk score is more strong associated than genome-wide score with Alzheimer's disease diagnosis in a case-control analysis. medRxiv https://doi.org/10.1101/2021.04.29.21256279 (2021).
Nho, K. et al. Association of Altered Liver Enzymes With Alzheimer Disease Diagnosis, Cognition, Neuroimaging Measures, and Cerebrospinal Fluid Biomarkers. JAMA Netw. Open 2, e197978-e197978 (2019).
Artikel PubMed PubMed Central Google Scholar
Reynolds, R. H. et al. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson's disease heritability. npj Park. Dis. 5, 1-14 (2019).
Artikel Google Scholar
Langston, R. G. et al. Association of a Common Genetic Variant with Parkinson's Disease is Propagated through Microglia. bioRxiv https://doi.org/10.1101/2021.01.15.426824 (2021).
Coetzee, S. G. et al. Enrichment of risk SNPs in regulatory regions implicate various tissues in Parkinson's disease etiology. Sci. Rep. 6, 1-11 (2016).
Artikel CAS Google Scholar
Schierding, W. et al. Common Variants Coregulate Expression of GBA and Modifier Genes to Delay Parkinson's Disease Onset. Mov. Disord. 35, 1346-1356 (2020).
CAS Artikel PubMed PubMed Central Google Scholar
Fasano, A., Visanji, N. P., Liu, L. W. C., Lang, A. E. & Pfeiffer, R. F. Gastrointestinale Dysfunktion bei der Parkinsonschen Krankheit. Lancet Neurol. 14, 625-639 (2015).
CAS Artikel PubMed Google Scholar
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291-295 (2015).
CAS Artikel PubMed PubMed Central Google Scholar
Engelender, S. & Isacson, O. The Threshold Theory for Parkinson's Disease. Trends Neurosci. 40, 4-14 (2017).
CAS Artikel PubMed Google Scholar
Andersen, M. S. et al. Heritability Enrichment Implicates Microglia in Parkinson's Disease Pathogenesis. Ann. Neurol. 89, 942-951 (2021).
Artikel PubMed Google Scholar
Bryois, J. et al. Genetische Identifizierung von Zelltypen, die den Merkmalen des Gehirnkomplexes zugrunde liegen, gibt Einblicke in die Ätiologie der Parkinson-Krankheit. Nat. Genet. 52, 482-493 (2020).
CAS Artikel PubMed PubMed Central Google Scholar
Booms, A., Pierce, S. E. & Coetzee, G. A. Parkinsons disease genetic risk evaluation in microglia highlights autophagy and lysosomal genes. bioRxiv 2020.08.17.254276 https://doi.org/10.1101/2020.08.17.254276 (2020).
Senkevich, K. & Gan-Or, Z. Autophagy lysosomal pathway dysfunction in Parkinson's disease; evidence from human genetics. Park. Relat. Disord. 73, 60-71 (2019).
Artikel Google Scholar
Stuart, T. & Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 20, 257-272 (2019).
CAS Artikel PubMed Google Scholar
Aldridge, S. & Teichmann, S. A. Single cell transcriptomics comes of age. Nat. Commun. 11, 1-4 (2020).
Artikel CAS Google Scholar
Przytycki, P. F. & Pollard, K. S. CellWalker integriert Einzelzell- und Massendaten, um regulatorische Elemente über Zelltypen hinweg in komplexen Geweben aufzulösen. Genome Biol. 22, 1-16 (2021).
Artikel CAS Google Scholar
Rizzo, G. et al. Genauigkeit der klinischen Diagnose der Parkinson-Krankheit. Neurology 86, 566-576 (2016).
Artikel PubMed Google Scholar
Malek, N. et al. Features of GBA-associated Parkinson's disease at presentation in the UK Tracking Parkinson's study. J. Neurol. Neurosurg. Psychiatry 89, 702-709 (2018).
Artikel PubMed Google Scholar
Gan-Or, Z. et al. LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 11, 121-125 (2010).
CAS Artikel PubMed Google Scholar
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177-1186 (2017).
CAS Artikel PubMed PubMed Central Google Scholar
Barton, N. H., Etheridge, A. M. & Véber, A. The infinitesimal model: Definition, Herleitung und Implikationen. Theor. Popul. Biol. 118, 50-73 (2017).
CAS Artikel PubMed Google Scholar
Barabási, A. L., Gulbahce, N. & Loscalzo, J. Network medicine: Ein netzwerkbasierter Ansatz für menschliche Krankheiten. Nat. Rev. Genet. 12, 56-68 (2011).
Artikel CAS PubMed PubMed Central Google Scholar
Lee, L. Y., Pandey, A. K., Maron, B. A. & Loscalzo, J. Network medicine in Cardiovascular Research. Cardiovasc. Res. 117, 2186-2202 (2020).
Artikel CAS PubMed Central Google Scholar
Ozturk, K., Dow, M., Carlin, D. E., Bejar, R. & Carter, H. The Emerging Potential for Network Analysis to Inform Precision Cancer Medicine. J. Mol. Biol. 430, 2875-2899 (2018).
CAS Artikel PubMed PubMed Central Google Scholar
Preston, J., VanZeeland, A. & Peiffer, D. A. Innovation at Illumina: The road to the $600 human genome. Nat. Portf. (2021).
Gasperini, M. et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens HHS Public Access. Cell 176, 377-390 (2019).
CAS Artikel PubMed PubMed Central Google Scholar
Tewhey, R. et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519-1529 (2016).
CAS Artikel PubMed PubMed Central Google Scholar
Fowler, D. M. et al. The Atlas of Variant Effects (AVE) Alliance: understanding genetic variation at nucleotide resolution. Zenodo (2021).
DeGeurin, M. Wearables saw explosive-but conditional-growth in 2020 - Insider Intelligence Trends, Forecasts & Statistics. eMarketer https://www.emarketer.com/content/wearables-saw-explosive-conditional-growth-2020 (2021).
Dunn, J. et al. Wearable sensors enable personalized predictions of clinical laboratory measurements. Nat. Med. 27, 1105-1112 (2021).
CAS Artikel PubMed PubMed Central Google Scholar
Sheehan, P. et al. PR001 Gentherapie verbessert Phänotypen in Modellen der Parkinson-Krankheit mit GBA1-Mutation. Alzheimer's Dement 16, e043614 (2020).
Google Scholar
Skrahina, V. et al. The Rostock International Parkinson's Disease (ROPAD) Study: Protocol and Initial Findings. Mov. Disord. 36, 1005-1010 (2021).
CAS Artikel PubMed Google Scholar
Schneider, S. A. & Alcalay, R. N. Precision medicine in Parkinson's disease: emerging treatments for genetic Parkinson's disease. J. Neurol. 267, 860-869 (2020).
CAS Artikel PubMed PubMed Central Google Scholar
Pushpakom, S. et al. Drug repurposing: Fortschritte, Herausforderungen und Empfehlungen. Nat. Rev. Drug Discov. 18, 41-58 (2018).
Artikel CAS PubMed Google Scholar
Toffoli, M. et al. Intronic Haplotypes in the GBA Gene Do Not Predict Age at Diagnosis of Parkinson's Disease. Mov. Disord. mds.28616 https://doi.org/10.1002/mds.28616 (2021).
Noyce, A. J. et al. PREDICT-PD: Identifizierung des Risikos für die Parkinson-Krankheit in der Bevölkerung: Methoden und Ausgangsergebnisse. J. Neurol. Neurosurg. Psychiatry 85, 31-37 (2014).
Artikel PubMed Google Scholar
Sturchio, A. et al. Phenotype-Agnostic Molecular Subtyping of Neurodegenerative Disorders: The Cincinnati Cohort Biomarker Program (CCBP). Front. Aging Neurosci. 12, 324 (2020).
Artikel CAS Google Scholar
Lillie, E. O. et al. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per. Med 8, 161-173 (2011).
Artikel PubMed PubMed Central Google Scholar
Schork, N. J. Personalisierte Medizin: Zeit für Ein-Personen-Studien. Nature 520, 609-611 (2015).
CAS Artikel PubMed Google Scholar
Margolis, A. & Giuliano, C. Making the switch: Von Fallstudien zu N-of-1-Studien. Epilepsy Behav. Rep. 12, 100336 (2019).
Artikel PubMed PubMed Central Google Scholar
Patrick, K. L., Bell, S. L., Weindel, C. G. & Watson, R. O. Exploring the 'multiple-hit hypothesis' of neurodegenerative disease: Bakterielle Infektionen sind der Schlüssel zum Erfolg. Front. Cell. Infect. Microbiol. 9, 1-18 (2019).
Artikel CAS Google Scholar
Riggare, S. & Hägglund, M. Precision Medicine in Parkinson's Disease - Exploring Patient-Initiated Self-Tracking. J. Parkinsons. Dis. 8, 441-446 (2018).
Artikel PubMed PubMed Central Google Scholar Referenzen herunterladen
Danksagung
S.F., A.C. und J.M.O’S. wurden von der Michael J. Fox Foundation for Parkinson’s Research und der Silverstein Foundation for Parkinson’s mit dem GBA-Zuschuss ID 16229 für J.M.O’S. finanziert. S.F. und J.M.O’S. wurden von der Neurological Foundation – Grant ID 3721588 (2008 SPG) – finanziert. S.F. wurde durch den Dines Family Charitable Trust finanziert. A.C. erhielt Zuschüsse von der australischen Regierung.
Angaben zum Autor
Zugehörigkeiten
Liggins Institute, Universität von Auckland, Auckland, Neuseeland
Sophie L. Farrow & Justin M. O'Sullivan
Maurice Wilkins Centre, Universität von Auckland, Auckland, Neuseeland
Sophie L. Farrow & Justin M. O'Sullivan
Australian Parkinson's Mission, Garvan Institute of Medical Research, Sydney, New South Wales, Australien
Antony A. Cooper & Justin M. O'Sullivan
Schule für klinische Medizin, UNSW Sydney, Sydney, New South Wales, Australien
Antony A. Cooper
MRC Lifecourse Epidemiology Unit, Universität von Southampton, Southampton, Vereinigtes Königreich
Justin M. O'SullivanAutoren
Sophie L. Farrow
Sie können auch in PubMed Google Scholar nach diesem Autor suchen
Antony A. Cooper
Sie können auch nach diesem Autor in PubMed Google Scholar suchen
Justin M. O'Sullivan
Sie können auch nach diesem Autor in PubMed Google Scholar suchenBeiträge
S.F. konzipierte und schrieb den ersten Entwurf und überarbeitete die nachfolgenden Entwürfe. A.C. und J.M.O’S. trugen zur Konzeption, kritischen Überprüfung und Überarbeitung der Arbeit bei. Alle Autoren genehmigten den endgültigen Entwurf zur Einreichung.
Korrespondierender Autor
Korrespondenz mit Justin M. O’Sullivan.
Ethische Erklärungen
Konkurrierende Interessen
Die Autoren erklären, dass sie keine konkurrierenden Interessen haben.
Zusätzliche Informationen
Anmerkung des Herausgebers: Springer Nature bleibt neutral in Bezug auf juristische Ansprüche in veröffentlichten Karten und institutionelle Zugehörigkeiten.
Ergänzende Informationen
Rechte und Genehmigungen
Open Access
Dieser Artikel steht unter einer Creative Commons Attribution 4.0 International License, die die Nutzung, Weitergabe, Anpassung, Verbreitung und Vervielfältigung in jedem Medium oder Format erlaubt, solange Sie den/die Originalautor(en) und die Quelle in angemessener Weise nennen, einen Link zur Creative Commons Lizenz angeben und angeben, ob Änderungen vorgenommen wurden.
Die Bilder oder anderes Material von Dritten in diesem Artikel sind in der Creative-Commons-Lizenz des Artikels enthalten, es sei denn, es wird in einer Kreditlinie zu dem Material anders angegeben. Wenn das Material nicht in der Creative-Commons-Lizenz des Artikels enthalten ist und die von Ihnen beabsichtigte Nutzung nicht durch gesetzliche Bestimmungen erlaubt ist oder über die erlaubte Nutzung hinausgeht, müssen Sie die Erlaubnis direkt beim Urheberrechtsinhaber einholen.
Eine Kopie dieser Lizenz können Sie unter http://creativecommons.org/licenses/by/4.0/ einsehen.
Nachdrucke und Genehmigungen
Über diesen Artikel
Überprüfen Sie die Aktualität und Authentizität über CrossMark
Diesen Artikel zitieren
Farrow, S.L., Cooper, A.A. & O’Sullivan, J.M. Redefining the hypotheses driving Parkinson’s diseases research. npj Parkinsons Dis. 8, 45 (2022). https://doi.org/10.1038/s41531-022-00307-w
Zitat herunterladen
Empfangen: 03. August 2021
Angenommen: 04 März 2022
Veröffentlicht: 19 April 2022
DOI: https://doi.org/10.1038/s41531-022-00307-wThemen
Redefining the hypotheses driving Parkinson’s diseases research
O’Sullivan, Justin M.49-62 Minuten
Abstract
Parkinson’s disease (PD) research has largely focused on the disease as a single entity centred on the development of neuronal pathology within the central nervous system. However, there is growing recognition that PD is not a single entity but instead reflects multiple diseases, in which different combinations of environmental, genetic and potential comorbid factors interact to direct individual disease trajectories. Moreover, an increasing body of recent research implicates peripheral tissues and non-neuronal cell types in the development of PD. These observations are consistent with the hypothesis that the initial causative changes for PD development need not occur in the central nervous system. Here, we discuss how the use of neuronal pathology as a shared, qualitative phenotype minimises insights into the possibility of multiple origins and aetiologies of PD. Furthermore, we discuss how considering PD as a single entity potentially impairs our understanding of the causative molecular mechanisms, approaches for patient stratification, identification of biomarkers, and the development of therapeutic approaches to PD. The clear consequence of there being distinct diseases that collectively form PD, is that there is no single biomarker or treatment for PD development or progression. We propose that diagnosis should shift away from the clinical definitions, towards biologically defined diseases that collectively form PD, to enable informative patient stratification. N-of-one type, clinical designs offer an unbiased, and agnostic approach to re-defining PD in terms of a group of many individual diseases.
Introduction
There is growing recognition that Parkinson’s disease is not a single entity1,2. Rather there are multiple different clinical, genetic and epidemiologically heterogeneous diseases that together are recognised within the one umbrella term of Parkinson’s disease3,4,5. Hereafter we refer to the multiple diseases as ‘PD’ for simplicity, and to prevent clouding the literature with a new term. Despite growing recognition of this concept, the majority of PD targeted research focuses on the ‘common’-pathological end-point of a linear PD storyline6: the physical manifestation of neuronal inclusions termed Lewy bodies, and the loss of dopaminergic neurons (DAn) within the central nervous system (CNS). This focus on the end-point pathology has proven its worth in the development of effective symptomatic therapies that include Levodopa7. However, the failure of nineteen phase 3 intervention trials8 targeting modification of disease progression illustrates a limitation of this focus. The restricted focus on endpoint pathology largely arises from issues including that PD diagnosis typically occurs many years after disease onset, predominantly on the basis of motor symptoms, and yet one can only study PD patients after this clinical diagnosis is made. The successful development of disease-modifying therapeutics has been further hindered by the absence of biomarkers, and more critically—the absence of informative, molecular mechanisms that define each of the individual diseases that collectively form PD. This is reflected in a lack of PD intervention trials that target specific mechanistic changes in groups of individual patients defined according to the mechanism(s) that contribute to disease development/progression. The SURE-PD3 trial is an exception that targeted only individuals with low serum urate concentrations9. However, beyond the SURE-PD3 trial, there is typically no specific measurable biological signal for the success of a disease-modifying intervention for each disease within the PD umbrella10. Instead, we remain reliant on relatively insensitive and variable clinical measures of PD progression8.
The advent of genome-wide association studies (GWAS) has enabled the identification of variants associated with risk of disease development11, different rates of cognitive decline12, and different rates of progression for PD13,14. However, the conglomeration of datasets needed to achieve the sample size and statistical power required for GWAS perpetuates the one-disease model of PD, and overlooks the presence of multiple different clinical, genetic and epidemiologically heterogeneous diseases. In these situations, the conglomeration of data across multiple different Parkinson diseases dilutes the frequency of specific disease-associated variants and thus reduces the ability to identify those variants that contribute to the trajectory of each individual disease (i.e., the aetiology). As such, the integration of genetic and standardised clinical data into a coherent coordinated approach to slow or prevent PD development, is yet to materialise. Achieving this requires that we move away from a dependence on the shared terminal pathology and clinical definitions and develop a means for patient stratification, using specific genetic information, that is based upon a sound understanding of the aetiology of each contributing molecular disease. But how can you achieve this, when to study the different diseases you must first define them? Here we will discuss how this circular argument can be broken using genetic, molecular and clinical information to identify the different trajectories within PD, from a prospective, disease risk-driven perspective, that stratifies patients and therapeutics without a priori assumptions.
Multiple disease trajectories beneath the Parkinson’s disease umbrella
In 2008, William Weiner wrote “there is no Parkinson disease”1 and suggested Parkinson diseases as a more fitting term for the observed multiple aetiologies. The term Parkinson diseases is consistent with the fact that there is no obvious, predictable disease trajectory following diagnosis, even in monogenic forms of the disease. Rather, each individual’s pathway is unique, or at most shared with a limited number of fellow patients15.
To illustrate the impact that treating PD as a group of diseases with different but overlapping aetiologies4,5,16 can have on our understanding of the disease, let us consider a conceptual model where each disease within PD is represented by a mountain within a range of mountains (Fig. 1). At present we are unable to accurately define the number of different diseases that collectively form PD, thus we have limited our model to seven mountains, for simplicity. In the PD mountain range model, an individual’s genetic risk is represented by the position in the valley (i.e., basecamp) where the individual starts climbing—this position naturally limits the mountain(s) that can be ascended and the route(s) that can be taken. PD patients cluster according to their basecamp, of which there are a limited number, defined by the potential and realised combinations of the risk variants within the genome. Environmental signals from the dynamic basecamp surroundings interact with the individual’s genetic factors to alter aspects of the disease, including onset age at which the patient begins climbing, or whether the individual even develops PD. These environmental signals include, among others: pesticides and pollutants17,18, diet19, viral infection20, head trauma21, inflammatory diseases22 (for an in-depth review on the role of environmental signals in relation to PD genetics see Johnson et al.23). Once an individual has begun ascending a mountain, the topology of the mountain, which represents the intrinsic (e.g. the gut microbiome24 or comorbid disease pathology25) and extrinsic (e.g. exercise26, diet19, and periodic fasting27) factors, influences how quickly each individual climbs the route (i.e., the rate of disease progression), and thus the presentation and severity of symptoms15.
Individual diseases that together comprise PD are heterogeneous in and of themselves. This is represented in our model by the existence of multiple routes to each mountain summit. These routes are not independent, merging and diverging, meaning it is likely that individuals can switch between the routes dependent on their particular combination of intrinsic and extrinsic factors. Although heterogeneity likely exists within each disease, it would ideally be sufficiently homogeneous to provide a single therapeutic target for treatment development. Furthermore, each route has different markers, or checkpoints, at different stages—akin to the biomarkers that provide an unbiased snap-shot that can be used to track disease development within individual patients. It is important to note however that these ‘on route’ biomarkers will change over the course of the disease, and are likely to be influenced by the individual’s age, diet, and combination of predisposing comorbid diseases.
It can be argued that there are commonalities across individual diseases that contribute to PD (i.e., shared between the different mountains within the range). Treating these commonalities would provide treatment for a larger group of patients. This may be true. However, whilst potentially useful, treating these commonalities would have limited benefit, as the symptoms (e.g. resting tremor and bradykinesia) appear late in the disease course, and thus patients would be more disabled (closer to their respective summits) by the time the treatment is initiated. Notably, disease-modifying interventions that target PD based on this premise have yielded little success thus far.
Other models of PD have been presented before. Perhaps best known is William Langston’s elephant model28 which captures the idea of diverse symptomology but still presents PD as a single disease, or, elephant. In our model, the elephant would be represented as a single mountain within the PD mountain range. Thus Langston’s model does not capture the multiple diseases that collectively form PD, or the heterogeneity that is inherent to each disease.
Using ‘omics to inform origins and trajectories of Parkinson’s disease
It is the patient’s combination of genetic risk coding (e.g. LRRK2-G2019S or SNCA-A53T) and non-coding variants that initially “set the stage” and determine which basecamp and mountain an individual will start ascending in their journey towards PD. The application of GWAS to the study of PD enables unbiased population-level identification of the genetic basis of risk that exist long before the disease initiates. However, the genetic variants that have been associated with PD by GWAS (e.g. 90 genetic loci11) only explain between 16-36% of the heritability of PD. Additionally, apart from a few exceptions, the odds ratios of the individual variants are typically low (e.g. between 0.8 – 1.2)11. Indeed, the current predictive ability of the SNPs associated with PD is so low as to make meaningful risk score prognosis unfeasible29. The missing heritability can partly be explained by issues with merging the multiple different diseases that contribute to PD, into the single entity that is defined by late-stage pathological markers (i.e., performing GWAS from the perspective of PD being a single disease). Furthermore, the reliance on a clinical definition means that no two ‘omics studies yield similar results since they only represent those of the heterogeneous patient population from which they were applied (e.g.30). Averaging these different but related datasets results in the identification of only the most significant risk loci that are common across all the diseases reaching statistical significance. The issue of averaging signals across the heterogeneous diseases that contribute to PD, when undertaking a GWAS, can be addressed by stratifying PD patients according to their genetically defined start-point, in turn enabling selection of informative longitudinal biomarkers and effective therapeutic approaches (specific to each route). This stratification can be achieved through genomic approaches that explore the specificities of GWAS manifestation31,32, and inform the distinct routes of PD development. As such, GWAS-based patient stratification could indicate 1) which pathway(s) is dysregulated; 2) pathway biomarkers to be examined; and 3) which targets should be considered for therapeutic intervention. However, shifting from simply identifying GWAS signals to informative stratification requires in depth characterisation of the causative variant(s) function33.
Until recently33, our inability to functionally translate non-coding genetic variation and risk to biologically disease-relevant pathways has meant that the earlier stages of PD development have been primarily neglected as a means of stratification or therapeutic intervention. In contrast to the noncoding risk variants, coding mutations in GBA and LRRK2 genes have been explored and enabled patients with these specific mutations to be stratified for therapeutic intervention, targeting these genetic subgroups of patients34,35. Furthermore, Szwedo et al. demonstrated a role for APOE-ε4 and GBA mutations in the rate of cognitive decline in PD patients, but found no significant impact for common variants in SNCA and MAPT12. These findings raise the possibility for earlier identification and stratification of individuals at high risk of rapid cognitive decline, thus highlighting suitable candidates for future targeted trials. Despite progress, the known incomplete penetrance of these mutations is problematic36 and highlights a remaining knowledge gap surrounding the mechanistic role of some of these mutations, such as the role of LRRK2 mutations in disease progression14. This therefore raises the question as to whether such interventions will be effective against disease progression even in patients with these specific mutations. Nonetheless, with recent advancements, our understanding of how both coding and non-coding risk manifests is evolving33,37. Such understanding can be used to inform hypotheses which will aid in the identification and stricter classification of individual diseases within PD that could also lead to targeted therapeutics.
Functional characterisation requires that the associated molecular, cellular and physiological phenotyping is sufficiently deep to allow accurate assignment of the causal variants and their target genes38, and potentially what tissue(s), the disease risk is conveyed in. Panyard et al. applied an approach to functionally characterise and assign the action of causal genetic variants in Alzheimer’s disease (AD)39. Briefly, Panyard et al. integrated genomic and clinical data from two longitudinal AD cohorts with epigenetic annotations to develop cell-type-specific genomic functional annotations39. These annotations were used to identify which SNPs are likely to be functional in different tissues39. The authors demonstrated that effects of these SNPs in the liver were statistically associated with Alzheimer’s diagnosis39. In so doing, Panyard et al. highlighted a potential contribution from the liver towards AD, including associations with core AD cerebrospinal-fluid biomarkers, in what is widely considered a ‘brain-centric’ disease. Whilst a small study (n = 79 AD patients), the finding that changes in the liver were predictive for some, but not all, individuals is consistent with the hypothesis that the liver malfunction accounts for one of the heterogeneous diseases that collectively contribute to AD40.
Genomic approaches are also being applied in attempts to identify and understand the cell- and tissue- types where genetic risk manifests in PD41,42. For example, Coetzee et al.43 used histone modification data combined with enrichment analyses to demonstrate that many PD-associated genetic variants were enriched, and had expression quantitative trait loci (eQTL) associations, in non-neuronal cell-types, including lymphocytes, mesendoderm-, liver- and adipocyte- cells43. Similarly, we have used a discovery-based approach to identify putative regulatory impacts of non-coding PD-associated risk variants in both the CNS and peripheral tissues33,44. Notably, our analyses indicated that eQTL effects for a subset (28%) of the 90 PD-associated risk variants were only detected in peripheral tissues (e.g. thyroid and oesophagus)33 while only 2% of PD risk SNPs had identifiable eQTLs solely in CNS tissues. Given that tissues are complex mixtures of cell types, the oesophageal finding does not imply that the effect is due to the muscles at the exclusion of the nerves that innervate the oesophagus. However, the finding is consistent with peripheral symptomology (e.g. dysphagia), that is sometimes observed in the early stages of PD45.
In an attempt to determine which tissues, and subsequently cell-types, are responsible for PD heritability, Reynolds et al.41 used stratified Linkage Disequilibrium score regression46 (see box 1) to measure the contribution(s) that common genetic variation makes to the heritability of PD across 53 tissues (inc. 13 brain region tissues), using schizophrenia as a comparative measure. In contrast to schizophrenia in which all 13 brain tissues were significantly enriched for heritability, there was no enrichment for PD heritability across any of the 53 tissues (in the CNS or peripheral tissues). The lack of PD heritability enrichment across these bulk tissues led Reynolds et al. to question whether cellular heterogeneity within tissues may be masking signals, and thus sought to investigate cell-type-specific enrichment of heritability. However, across 6 human and 30 mouse CNS cell-types, Reynolds et al. identified no cell-type enrichment for PD heritability. The Lewy Body pathology in specific neuronal cell types, associated with PD, has encouraged researchers to focus efforts towards understanding risk in neuronal subtypes. However, the findings from Reynolds et al. provide reason to believe that risk loci are affecting non-CNS cell-types and/or cellular processes and pathways across multiple cell types, and to which different cell types have varying vulnerability41. Such varying vulnerability, consistent with the proposed threshold theory for PD47, could likely be a result of interactions with environmental factors and/or comorbid disease pathology.
In contrast to the lack of cell-type heritability enrichment identified by Reynolds et al., there have been multiple studies to date that implicate glial cell types, mostly microglia, in neuroinflammation and PD pathogenesis42,48,49. Given these implications, Bryois et al. combined cell-type-specific gene expression and GWAS data to explore the role of glial cells in PD pathogenesis49. Roles for microglia were indicated by the finding that cell-type-specific ATACseq identified functional PD risk loci that were enriched for autophagy and lysosomal processes50, both of which have been previously implicated in PD51. Furthermore, elevated LRRK2 expression, associated with the linked PD GWAS SNPs rs76904798 and rs7294619 (R2 = 0.842), has also been shown to occur specifically in microglia42. Collectively, these data are consistent with the hypothesis that PD genetic risk variants affect non-neuronal cell types of the CNS. However, while these studies highlight the importance of cell-type consideration, they are still driven by a priori assumptions that are CNS focused. As such, it is essential to extend these analyses to non-CNS cell-types, following a more discovery-based, hypothesis-free approach, to determine if such risk enrichment is truly specific to the microglia, or if other non-CNS cell-types may also be involved in disease initiation and propagation.
Together these studies highlight how multiple ‘omics approaches can be used to identify the tissue- and cell-type-specific manifestations for GWAS risk variants. The findings we have discussed support two potential, non-mutually exclusive, hypotheses: First, the individual diseases within the PD umbrella may arise through genetic variation-dependent mechanisms that dictate the tissue-of-origin(s) and thus the pathological pathways associated with the disease. This concept is reflected in the mountain range model, with each basecamp representing a different, genetically-informed, start-point. In the second hypothesis, variants impacting a specific peripheral tissue- or cell-type, cause dysregulation that adds to the disease complexity/symptoms without necessarily leading to the CNS pathology that is typically associated with PD. This second hypothesis aligns with the threshold theory for PD which was developed on the basis of parallel degeneration of both the central and peripheral nervous systems47. As such, there is a need to look beyond the tissue- and cell-types that are traditionally associated with PD pathology to gain a greater understanding of the mechanisms through which genetic risk may be manifested. Advances within the fields of single-cell transcriptomics52,53 and bulk-cell analyses54 will provide additional insights that begin to untangle the relative contributions of genetics and the environment to PD risk manifestation. But the question remains, how do we apply these approaches to a mechanistically-heterogeneous disease?
Using big data to identify individual trajectories in a heterogeneous disease
Conglomerating data from different cohorts provides a large sample size (n) which is otherwise unachievable from a single-centre cohort. As such, conglomerated data provides much-needed statistical power to address particular hypotheses. Despite providing statistical power, the conglomeration of different PD cohorts unfortunately also highlights the lack of strict diagnostic criteria for PD and related diseases, with different cohorts often using different diagnostic criteria30. A further confounding problem, that affects diagnosis even at the level of a single clinician, is misclassification55. Such misclassification raises the problem of inclusion of non-PD patients in cohorts, which may be skewing outcomes of observational studies and clinical trials. A third, and substantial, complicating factor is the likely multiple different mechanistic diseases that exist within the ‘homogenous’ clinical PD cohorts currently studied. This problem is particularly prevalent in cohorts that include patients with different genetic predispositions to diseases within PD, such as GBA-PD and LRRK2-PD patients, who typically present with different symptomatic trajectories56,57. Grouping these different individual diseases together is likely causing a loss of information. If data conglomeration is to achieve what is hoped, disease biomarkers, and more specifically biomarkers for the different diseases that collectively form PD are urgently needed. The need to define individual diseases as opposed to merging them into a single entity is in line with the prediction made by Espay and Lang that smaller, smarter clinical trials are needed to move away from this ‘homogeneous’ clinical Parkinson’s phenotype6.
As discussed earlier, genetic risk variants offer an option for such genetic stratification—with an individual’s risk profile determining their disease starting point (e.g. specific basecamp in the mountain range model). These genetic risk variants, or SNPs, do not however act independently33. Rather they act in a combinatorial manner within a much larger genetic background. In order to understand the full contribution that PD-associated SNPs make to PD, they need to be considered in the context of the omnigenic58 and infinitesimal59 models for disease (see box 1), and in terms of network medicine60. Network medicine approaches enable the disease to be contextualised as a sum of inter-connected perturbations, reflective of the underlying genetic and molecular risk drivers (i.e. studying PD risk variants in the context of an individual’s complete genotype). The utility of network medicine60 has being explored in other complex diseases, and has already aided in the identification of novel targets for therapeutic strategies and development61,62.
Exploring the impact and interconnectivity of genetic contributions to an individual’s disease risk profile, from a network medicine angle, has only become feasible following recent advancements. These include the reductions in costs for genome sequencing and computing63, and the development of machine learning approaches to detect complex patterns in genomes. Such advances have informed, and been enhanced by, the rapidly evolving post-GWAS genome-editing toolbox, including CRISPR screens64 and massively parallel reporter assays65 (to test observed patterns for functional significance). These tools will over time provide the data required to understand the complete genetic contributions to the development of the diseases that collectively form PD, amongst other complex diseases. Collaborative efforts, such as the Atlas of Variant Effects Alliance (https://www.varianteffect.org/)66, will be critical in enabling the curation and systematic collation of results from these functional post-GWAS studies. Another recent technological advance that will likely enhance genomic findings from a phenotypic perspective is the introduction of wearables67. Such devices have been shown to provide vital sign data (e.g. heart rate and electrodermal activity) at a level equivalent to that gained in a clinical setting68. The widespread uptake of these wearables enables individualised, longitudinal and continuous health monitoring. While identifying signal from noise in movement measurements is challenging, combining the in-depth phenotypic data that wearables provide with matched genetic data promises to aid in identifying clinical differences amongst the different genomic diseases within PD.
Information on genetic variation and drug responses can be used to help determine which drugs, are likely to be safe and efficacious in an individual. These approaches are leading to the emergence of ‘genetically-informed’ clinical trials (i.e., precision medicine approaches) in PD69,70,71. For example, Ambroxol has been repurposed to treat PD patients with a GBA coding mutation34. Despite having only been trialled in a small, open label, non-randomised group of individuals, Ambroxol shows promise for the treatment of this well-defined yet heterogeneous (i.e., it included multiple GBA coding mutations) subset of individuals34. The Ambroxol trial is an exemplar that paves the way for future precision-informed clinical trials in PD. Not only does it address the issue of treating patients according to genomic information, but also shows the potential of repurposing already licensed medication72, to accelerate the process of drug development. The Ambroxol trial also included some idiopathic PD patients—of whom also showed promising responses to the treatment. Identifying idiopathic PD patients who specifically have reduced GCase activity (i.e., those with GBA modifying genotypes30,44) may lead to better outcomes for patients.
Despite the obvious promise of a stratified approach to clinical testing and therapy, the lack of genotyping as a part of clinical assessment means that the identification of the relatively small numbers of individuals with genetic predispositions remains a major financial and temporal challenge. However, this is changing as initiatives, such as PD frontline (https://pdfrontline.com/en) and PD GENEration (https://www.parkinson.org/PDGENEration), are offering genetic testing for PD patients to ensure individuals carrying defined mutations are referred to the clinical trials best suited to them.
Concluding remarks & future perspectives
Recognising that many diseases contribute to PD highlights a challenge that is present in the search for a biomarker of PD progression and therapeutics. Specifically, if there are many diseases subsumed within the umbrella of PD, then we should be looking for biomarkers for each individual disease. That we continue selecting patients on the basis of clinical criteria rather than biological ones impairs our ability to do this. Even genetic risk for PD is currently viewed within the context of the shared pathology that connects the different Parkinson diseases. The utility of network medicine60 has been established in other complex diseases, aiding the identification of novel targets for therapeutic strategies and development61,62. While it is certainly true that further initiatives involving large-scale data conglomeration will aid in the molecular and clinical understanding of the disease, the lack of uniformity in PD diagnosis and disease trajectories will likely confound findings from genomic and biomarker studies30,73. Recent initiatives (e.g. PREDICT-PD74 and the Cincinnati Cohort Biomarker Program (CCBP)75) that incorporate discovery-based analyses of prospective cohorts are seeking to address this by defining PD developmental pathways and biomarkers. Furthermore, we contend that it is time to consider systematic n-of-176,77,78 approaches (see box 1) in PD research, to identify the combinations and relative contributions of the genetic, pathological and environmental factors in each unique circumstance3,79, for individuals within a heterogeneous population. The population’s use of wearables will contribute to the collection of relevant data for achieving such an approach80. Ultimately, the aggregated results of n-of-1 approaches will help elucidate the many diseases that contribute to the one complex Parkinson disease. Redefining the hypotheses driving PD research will enable movement away from the current focus on shared pathology and clinical definitions. This in turn will make way for the development of targeted diagnostic and therapeutic approaches that are based upon a molecular understanding of the aetiology of the individual diseases, and thus have the ability to slow, stop or reverse disease progression and ultimately achieve disease prevention.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
There is no data to share that is specific to this perspective paper. All information is included in the references.
References
- Weiner, W. J. There is no Parkinson disease. Arch. Neurol. 65, 705–708 (2008).Article PubMed Google Scholar
- Espay, A. J. et al. Disease modification and biomarker development in Parkinson disease. Neurology 94, 481–494 (2020).Article PubMed PubMed Central Google Scholar
- Mestre, T. A. et al. Parkinson’s Disease Subtypes: Critical Appraisal and Recommendations. J. Parkinsons. Dis.11, 395–404 (2021).Article PubMed PubMed Central Google Scholar
- Espay, A. J., Brundin, P. & Lang, A. E. Precision medicine for disease modification in Parkinson disease. Nat. Rev. Neurol. 13, 119–126 (2017).Article PubMed Google Scholar
- Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet 397, 2284–2303 (2021).CAS Article PubMed Google Scholar
- Espay, A. J. & Lang, A. E. Parkinson diseases in the 2020s and beyond: Replacing clinico-pathologic convergence with systems biology divergence. J. Parkinsons. Dis. 8, S59–S64 (2018).Article PubMed PubMed Central Google Scholar
- Paoletti, F. P., Tambasco, N. & Parnetti, L. Levodopa treatment in Parkinson’s disease: earlier or later? Ann. Transl. Med. 7, S189–S189 (2019).Article Google Scholar
- Espay, A. J. & Stecher, B. Brain Fables: The hidden history of neurodegenerative diseases and a blueprint to conquer them. https://doi.org/10.1017/9781108888202 (Cambridge University Press, 2020).
- Schwarzschild, M. A. et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 326, 926–939 (2021).CAS Article PubMed Google Scholar
- Espay, A. J. et al. Biomarker‐driven phenotyping in Parkinson’s disease: A translational missing link in disease‐modifying clinical trials. Mov. Disord. 32, 319–324 (2017).Article PubMed PubMed Central Google Scholar
- Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).CAS Article PubMed PubMed Central Google Scholar
- Szwedo, A. A. et al. GBA and APOE Impact Cognitive Decline in Parkinson’s Disease: A 10-Year Population-Based Study. Mov. Disord. https://doi.org/10.1002/MDS.28932 (2022).
- Tan, M. M. X. et al. Genome‐Wide Association Studies of Cognitive and Motor Progression in Parkinson’s Disease. Mov. Disord. 36, 424–433 (2020).Article CAS PubMed Google Scholar
- Liu, G. et al. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat. Genet. 53, 787–793 (2021).CAS Article PubMed PubMed Central Google Scholar
- Ravan, A. et al. Non-motor symptoms in an Indian cohort of Parkinson’s disease patients and correlation of progression of non-motor symptoms with motor worsening. Neurol. India 63, 166 (2015).Article PubMed Google Scholar
- Berg, D. et al. Prodromal Parkinson disease subtypes — key to understanding heterogeneity. Nat. Rev. Neurol.17, 349–361 (2021).Article PubMed Google Scholar
- Pezzoli, G. & Cereda, E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 80, 2035–2041 (2013).CAS Article PubMed Google Scholar
- Kamel, F. Paths from pesticides to Parkinson’s. Science 341, 722–723 (2013).CAS Article PubMed Google Scholar
- Paknahad, Z., Sheklabadi, E., Derakhshan, Y., Bagherniya, M. & Chitsaz, A. The effect of the Mediterranean diet on cognitive function in patients with Parkinson’s disease: A randomized clinical controlled trial. Complement. Ther. Med. 50, 102366 (2020).Article PubMed Google Scholar
- Olsen, L. K., Dowd, E. & McKernan, D. P. A role for viral infections in Parkinson’s etiology? Neuronal Signal 2, 20170166 (2018).Article Google Scholar
- Paul, K. C. et al. The association between lifestyle factors and Parkinson’s disease progression and mortality. Mov. Disord. 34, 58–66 (2019).Article PubMed PubMed Central Google Scholar
- Villumsen, M., Aznar, S., Pakkenberg, B., Jess, T. & Brudek, T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut 68, 18–24 (2019).CAS Article PubMed Google Scholar
- Johnson, M. E., Stecher, B., Labrie, V., Brundin, L. & Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 42, 4–13 (2019).CAS Article PubMed Google Scholar
- Heinzel, S. et al. Gut Microbiome Signatures of Risk and Prodromal Markers of Parkinson Disease. Ann. Neurol.88, 320–331 (2020).Article PubMed Google Scholar
- Cheong, J. L. Y., de Pablo-Fernandez, E., Foltynie, T. & Noyce, A. J. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Parkinsons. Dis. 10, 775–789 (2020).CAS Article PubMed PubMed Central Google Scholar
- Crotty, G. F. & Schwarzschild, M. A. Chasing Protection in Parkinson’s Disease: Does Exercise Reduce Risk and Progression? Front. Aging Neurosci. 12, 1–11 (2020).Article Google Scholar
- Neth, B. J., Bauer, B. A., Benarroch, E. E. & Savica, R. The Role of Intermittent Fasting in Parkinson’s Disease. Front. Neurol. 12, 1–7 (2021).Article Google Scholar
- Langston, J. W. The Parkinson’s complex: Parkinsonism is just the tip of the Iceberg. Ann. Neurol. 59, 591–596 (2006).Article PubMed Google Scholar
- Koch, S. et al. Validity and Prognostic Value of a Polygenic Risk Score for Parkinson’s Disease. Genes (Basel)12, 1859 (2021).CAS Article Google Scholar
- O’Sullivan, J. M., Heijer, J. M., Groeneveld, G. J. & Cooper, A. A. Intronic Haplotypes in GBA Modify Age at Diagnosis of Parkinson’s: Replication in a Subgroup. Mov. Disord. 36, 1468–1470 (2021).Article PubMed Google Scholar
- Novikova, G. et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 12, 1–14 (2021).Article CAS Google Scholar
- Wang, Q. et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 10, 1–15 (2019).Article CAS Google Scholar
- Farrow, S. L. et al. Establishing gene regulatory networks from Parkinson’s disease risk loci. Brain 139, 1–36 (2022).Google Scholar
- Mullin, S. et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations. JAMA Neurol. 77, 427–434 (2020).Article PubMed PubMed Central Google Scholar
- Ding, X. & Ren, F. Leucine-rich repeat kinase 2 inhibitors: a patent review (2014-present). Expert Opin. Ther. Pat. 30, 275–286 (2020).CAS Article PubMed Google Scholar
- Anheim, M. et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78, 417–420 (2012).CAS Article PubMed Google Scholar
- Ho, D. et al. Machine Learning Identifies Six Genetic Variants and Alterations in the Heart Atrial Appendage as Key Contributors to PD Risk Predictivity. Front. Genet. 12, 2409 (2022).Article Google Scholar
- Karthik Jagadeesh, A. A. et al. Identifying disease-critical cell types and cellular processes across the human body by integration of single-cell profiles and human genetics. bioRxiv 2021.03.19.436212 https://doi.org/10.1101/2021.03.19.436212 (2021).
- Panyard, D. J. et al. Liver-specific polygenic risk score is more strongly associated than genome-wide score with Alzheimer’s disease diagnosis in a case-control analysis. medRxiv https://doi.org/10.1101/2021.04.29.21256279(2021).
- Nho, K. et al. Association of Altered Liver Enzymes With Alzheimer Disease Diagnosis, Cognition, Neuroimaging Measures, and Cerebrospinal Fluid Biomarkers. JAMA Netw. Open 2, e197978–e197978 (2019).Article PubMed PubMed Central Google Scholar
- Reynolds, R. H. et al. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. npj Park. Dis. 5, 1–14 (2019).Article Google Scholar
- Langston, R. G. et al. Association of a Common Genetic Variant with Parkinson’s Disease is Propagated through Microglia. bioRxiv https://doi.org/10.1101/2021.01.15.426824 (2021).
- Coetzee, S. G. et al. Enrichment of risk SNPs in regulatory regions implicate diverse tissues in Parkinson’s disease etiology. Sci. Rep. 6, 1–11 (2016).Article CAS Google Scholar
- Schierding, W. et al. Common Variants Coregulate Expression of GBA and Modifier Genes to Delay Parkinson’s Disease Onset. Mov. Disord. 35, 1346–1356 (2020).CAS Article PubMed PubMed Central Google Scholar
- Fasano, A., Visanji, N. P., Liu, L. W. C., Lang, A. E. & Pfeiffer, R. F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 14, 625–639 (2015).CAS Article PubMed Google Scholar
- Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).CAS Article PubMed PubMed Central Google Scholar
- Engelender, S. & Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 40, 4–14 (2017).CAS Article PubMed Google Scholar
- Andersen, M. S. et al. Heritability Enrichment Implicates Microglia in Parkinson’s Disease Pathogenesis. Ann. Neurol. 89, 942–951 (2021).Article PubMed Google Scholar
- Bryois, J. et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 52, 482–493 (2020).CAS Article PubMed PubMed Central Google Scholar
- Booms, A., Pierce, S. E. & Coetzee, G. A. Parkinsons disease genetic risk evaluation in microglia highlights autophagy and lysosomal genes. bioRxiv 2020.08.17.254276 https://doi.org/10.1101/2020.08.17.254276 (2020).
- Senkevich, K. & Gan-Or, Z. Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Park. Relat. Disord. 73, 60–71 (2019).Article Google Scholar
- Stuart, T. & Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 20, 257–272 (2019).CAS Article PubMed Google Scholar
- Aldridge, S. & Teichmann, S. A. Single cell transcriptomics comes of age. Nat. Commun. 11, 1–4 (2020).Article CAS Google Scholar
- Przytycki, P. F. & Pollard, K. S. CellWalker integrates single-cell and bulk data to resolve regulatory elements across cell types in complex tissues. Genome Biol. 22, 1–16 (2021).Article CAS Google Scholar
- Rizzo, G. et al. Accuracy of clinical diagnosis of Parkinson disease. Neurology 86, 566–576 (2016).Article PubMed Google Scholar
- Malek, N. et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 89, 702–709 (2018).Article PubMed Google Scholar
- Gan-Or, Z. et al. LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 11, 121–125 (2010).CAS Article PubMed Google Scholar
- Boyle, E. A., Li, Y. I. & Pritchard, J. K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017).CAS Article PubMed PubMed Central Google Scholar
- Barton, N. H., Etheridge, A. M. & Véber, A. The infinitesimal model: Definition, derivation, and implications. Theor. Popul. Biol. 118, 50–73 (2017).CAS Article PubMed Google Scholar
- Barabási, A. L., Gulbahce, N. & Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 (2011).Article CAS PubMed PubMed Central Google Scholar
- Lee, L. Y., Pandey, A. K., Maron, B. A. & Loscalzo, J. Network medicine in Cardiovascular Research. Cardiovasc. Res. 117, 2186–2202 (2020).Article CAS PubMed Central Google Scholar
- Ozturk, K., Dow, M., Carlin, D. E., Bejar, R. & Carter, H. The Emerging Potential for Network Analysis to Inform Precision Cancer Medicine. J. Mol. Biol. 430, 2875–2899 (2018).CAS Article PubMed PubMed Central Google Scholar
- Preston, J., VanZeeland, A. & Peiffer, D. A. Innovation at Illumina: The road to the $600 human genome. Nat. Portf. (2021).
- Gasperini, M. et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens HHS Public Access. Cell 176, 377–390 (2019).CAS Article PubMed PubMed Central Google Scholar
- Tewhey, R. et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529 (2016).CAS Article PubMed PubMed Central Google Scholar
- Fowler, D. M. et al. The Atlas of Variant Effects (AVE) Alliance: understanding genetic variation at nucleotide resolution. Zenodo (2021).
- DeGeurin, M. Wearables saw explosive—but conditional—growth in 2020 – Insider Intelligence Trends, Forecasts & Statistics. eMarketer https://www.emarketer.com/content/wearables-saw-explosive-conditional-growth-2020 (2021).
- Dunn, J. et al. Wearable sensors enable personalized predictions of clinical laboratory measurements. Nat. Med.27, 1105–1112 (2021).CAS Article PubMed PubMed Central Google Scholar
- Sheehan, P. et al. PR001 gene therapy improved phenotypes in models of Parkinson’s disease with GBA1 mutation. Alzheimer’s Dement 16, e043614 (2020).Google Scholar
- Skrahina, V. et al. The Rostock International Parkinson’s Disease (ROPAD) Study: Protocol and Initial Findings. Mov. Disord. 36, 1005–1010 (2021).CAS Article PubMed Google Scholar
- Schneider, S. A. & Alcalay, R. N. Precision medicine in Parkinson’s disease: emerging treatments for genetic Parkinson’s disease. J. Neurol. 267, 860–869 (2020).CAS Article PubMed PubMed Central Google Scholar
- Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2018).Article CAS PubMed Google Scholar
- Toffoli, M. et al. Intronic Haplotypes in the GBA Gene Do Not Predict Age at Diagnosis of Parkinson’s Disease. Mov. Disord. mds.28616 https://doi.org/10.1002/mds.28616 (2021).
- Noyce, A. J. et al. PREDICT-PD: Identifying risk of Parkinson’s disease in the community: Methods and baseline results. J. Neurol. Neurosurg. Psychiatry 85, 31–37 (2014).Article PubMed Google Scholar
- Sturchio, A. et al. Phenotype-Agnostic Molecular Subtyping of Neurodegenerative Disorders: The Cincinnati Cohort Biomarker Program (CCBP). Front. Aging Neurosci. 12, 324 (2020).Article CAS Google Scholar
- Lillie, E. O. et al. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per. Med 8, 161–173 (2011).Article PubMed PubMed Central Google Scholar
- Schork, N. J. Personalized medicine: Time for one-person trials. Nature 520, 609–611 (2015).CAS Article PubMed Google Scholar
- Margolis, A. & Giuliano, C. Making the switch: From case studies to N-of-1 trials. Epilepsy Behav. Rep. 12, 100336 (2019).Article PubMed PubMed Central Google Scholar
- Patrick, K. L., Bell, S. L., Weindel, C. G. & Watson, R. O. Exploring the ‘multiple-hit hypothesis’ of neurodegenerative disease: Bacterial infection comes up to bat. Front. Cell. Infect. Microbiol. 9, 1–18 (2019).Article CAS Google Scholar
- Riggare, S. & Hägglund, M. Precision Medicine in Parkinson’s Disease – Exploring Patient-Initiated Self-Tracking. J. Parkinsons. Dis. 8, 441–446 (2018).Article PubMed PubMed Central Google Scholar
Acknowledgements
S.F., A.C., and J.M.O’S. were funded by the Michael J Fox Foundation for Parkinson’s research and the Silverstein Foundation for Parkinson’s with GBA—grant ID 16229 to J.M.O’S. S.F. and J.M.O’S. were funded by the Neurological Foundation—grant ID 3721588 (2008 SPG). S.F. was funded by the Dines Family Charitable Trust. A.C. received grant funding from the Australian Government.
Author information
Affiliations
- Liggins Institute, The University of Auckland, Auckland, New ZealandSophie L. Farrow & Justin M. O’Sullivan
- The Maurice Wilkins Centre, The University of Auckland, Auckland, New ZealandSophie L. Farrow & Justin M. O’Sullivan
- Australian Parkinson’s Mission, Garvan Institute of Medical Research, Sydney, New South Wales, AustraliaAntony A. Cooper & Justin M. O’Sullivan
- School of Clinical Medicine, UNSW Sydney, Sydney, New South Wales, AustraliaAntony A. Cooper
- MRC Lifecourse Epidemiology Unit, University of Southampton, Southampton, United KingdomJustin M. O’Sullivan
Authors
- Sophie L. FarrowYou can also search for this author in PubMed Google Scholar
- Antony A. CooperYou can also search for this author in PubMed Google Scholar
- Justin M. O’SullivanYou can also search for this author in PubMed Google Scholar
Contributions
S.F. conceived and wrote the first draft, and revised subsequent drafts. A.C. and J.M.O’S. contributed to conception, critical review, and revision of the paper. All authors approved the final draft for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Farrow, S.L., Cooper, A.A. & O’Sullivan, J.M. Redefining the hypotheses driving Parkinson’s diseases research. npj Parkinsons Dis. 8, 45 (2022). https://doi.org/10.1038/s41531-022-00307-w
- Received: 03 August 2021
- Accepted: 04 March 2022
- Published: 19 April 2022
- DOI: https://doi.org/10.1038/s41531-022-00307-w